How CRISPR Is Changing Cancer Research and Treatment
July 27, 2020 , by NCI Staff

CRISPR is a highly precise gene editing tool that is changing cancer research and treatment.
Ever since scientists realized that changes in DNA cause cancer , they have been searching for an easy way to correct those changes by manipulating DNA . Although several methods of gene editing have been developed over the years, none has really fit the bill for a quick, easy, and cheap technology.
But a game-changer occurred in 2013, when several researchers showed that a gene-editing tool called CRISPR could alter the DNA of human cells like a very precise and easy-to-use pair of scissors.
The new tool has taken the research world by storm, markedly shifting the line between possible and impossible. As soon as CRISPR made its way onto the shelves and freezers of labs around the world, cancer researchers jumped at the chance to use it.
“CRISPR is becoming a mainstream methodology used in many cancer biology studies because of the convenience of the technique,” said Jerry Li, M.D., Ph.D., of NCI’s Division of Cancer Biology .
Now CRISPR is moving out of lab dishes and into trials of people with cancer. In a small study, for example, researchers tested a cancer treatment involving immune cells that were CRISPR-edited to better hunt down and attack cancer.
Despite all the excitement, scientists have been proceeding cautiously, feeling out the tool’s strengths and pitfalls, setting best practices, and debating the social and ethical consequences of gene editing in humans.

How Does CRISPR Work?
Like many other advances in science and medicine, CRISPR was inspired by nature. In this case, the idea was borrowed from a simple defense mechanism found in some microbes, such as bacteria.
To protect themselves against invaders like viruses, these microbes capture snippets of the intruder’s DNA and store them away as segments called CRISPRs, or clustered regularly interspersed short palindromic repeats. If the same germ tries to attack again, those DNA segments (turned into short pieces of RNA ) help an enzyme called Cas find and slice up the invader’s DNA.
After this defense system was discovered, scientists realized that it had the makings of a versatile gene-editing tool. Within a handful of years, multiple groups had successfully adapted the system to edit virtually any section of DNA, first in the cells of other microbes, and then eventually in human cells.

CRISPR consists of a guide RNA (RNA-targeting device, purple) and the Cas enzyme (blue). When the guide RNA matches up with the target DNA (orange), Cas cuts the DNA. A new segment of DNA (green) can then be added.
In the laboratory, the CRISPR tool consists of two main actors: a guide RNA and a DNA-cutting enzyme, most commonly one called Cas9. Scientists design the guide RNA to mirror the DNA of the gene to be edited (called the target). The guide RNA partners with Cas and—true to its name—leads Cas to the target. When the guide RNA matches up with the target gene's DNA, Cas cuts the DNA.
What happens next depends on the type of CRISPR tool that’s being used. In some cases, the target gene's DNA is scrambled while it's repaired, and the gene is inactivated . With other versions of CRISPR, scientists can manipulate genes in more precise ways such as adding a new segment of DNA or editing single DNA letters .
Scientists have also used CRISPR to detect specific targets, such as DNA from cancer-causing viruses and RNA from cancer cells . Most recently, CRISPR has been put to use as an experimental test to detect the novel coronavirus .
Why Is CRISPR a Big Deal?
Scientists consider CRISPR to be a game-changer for a number of reasons. Perhaps the biggest is that CRISPR is easy to use, especially compared with older gene-editing tools.
“Before, only a handful of labs in the world could make the proper tools [for gene editing]. Now, even a high school student can make a change in a complex genome ” using CRISPR, said Alejandro Chavez, M.D., Ph.D., an assistant professor at Columbia University who has developed several novel CRISPR tools.
CRISPR is also completely customizable. It can edit virtually any segment of DNA within the 3 billion letters of the human genome, and it’s more precise than other DNA-editing tools.
And gene editing with CRISPR is a lot faster. With older methods, “it usually [took] a year or two to generate a genetically engineered mouse model , if you’re lucky,” said Dr. Li. But now with CRISPR, a scientist can create a complex mouse model within a few months, he said.
Another plus is that CRISPR can be easily scaled up. Researchers can use hundreds of guide RNAs to manipulate and evaluate hundreds or thousands of genes at a time. Cancer researchers often use this type of experiment to pick out genes that might make good drug targets .
And as an added bonus, “it’s certainly cheaper than previous methods,” Dr. Chavez noted.
What Are CRISPR’s Limitations?
With all of its advantages over other gene-editing tools, CRISPR has become a go-to for scientists studying cancer. There’s also hope that it will have a place in treating cancer, too. But CRISPR isn’t perfect, and its downsides have made many scientists cautious about its use in people.
A major pitfall is that CRISPR sometimes cuts DNA outside of the target gene—what’s known as “off-target” editing. Scientists are worried that such unintended edits could be harmful and could even turn cells cancerous , as occurred in a 2002 study of a gene therapy .
“If [CRISPR] starts breaking random parts of the genome, the cell can start stitching things together in really weird ways, and there’s some concern about that becoming cancer,” Dr. Chavez explained. But by tweaking the structures of Cas and the guide RNA, scientists have improved CRISPR’s ability to cut only the intended target, he added.
Another potential roadblock is getting CRISPR components into cells. The most common way to do this is to co-opt a virus to do the job. Instead of ferrying genes that cause disease, the virus is modified to carry genes for the guide RNA and Cas.
Slipping CRISPR into lab-grown cells is one thing; but getting it into cells in a person's body is another story. Some viruses used to carry CRISPR can infect multiple types of cells, so, for instance, they may end up editing muscle cells when the goal was to edit liver cells.
Researchers are exploring different ways to fine-tune the delivery of CRISPR to specific organs or cells in the human body. Some are testing viruses that infect only one organ, like the liver or brain. Others have created tiny structures called nanocapsules that are designed to deliver CRISPR components to specific cells.
Because CRISPR is just beginning to be tested in humans, there are also concerns about how the body—in particular, the immune system —will react to viruses carrying CRISPR or to the CRISPR components themselves.
Some wonder whether the immune system could attack Cas (a bacterial enzyme that is foreign to human bodies) and destroy CRISPR-edited cells. Twenty years ago, a patient died after his immune system launched a massive attack against the viruses carrying a gene therapy he had received. However, newer CRISPR-based approaches rely on viruses that appear to be safer than those used for older gene therapies.
Another major concern is that editing cells inside the body could accidentally make changes to sperm or egg cells that can be passed on to future generations. But for almost all ongoing human studies involving CRISPR, patients’ cells are removed and edited outside of their bodies. This “ ex vivo ” approach is considered safer because it is more controlled than trying to edit cells inside the body, Dr. Chavez said.
However, one ongoing study is testing CRISPR gene editing directly in the eyes of people with a genetic disease that causes blindness, called Leber congenital amaurosis.
The First Clinical Trial of CRISPR for Cancer
The first trial in the United States to test a CRISPR-made cancer therapy was launched in 2019 at the University of Pennsylvania. The study, funded in part by NCI, is testing a type of immunotherapy in which patients’ own immune cells are genetically modified to better “see” and kill their cancer.
The therapy involves making four genetic modifications to T cells , immune cells that can kill cancer. First, the addition of a synthetic gene gives the T cells a claw-like protein (called a receptor ) that “sees” NY-ESO-1, a molecule on some cancer cells.
Then CRISPR is used to remove three genes: two that can interfere with the NY-ESO-1 receptor and another that limits the cells’ cancer-killing abilities. The finished product, dubbed NYCE T cells, were grown in large numbers and then infused into patients.

The first trial of CRISPR for patients with cancer tested T cells that were modified to better "see" and kill cancer. CRISPR was used to remove three genes: two that can interfere with the NY-ESO-1 receptor and another that limits the cells’ cancer-killing abilities.
“We had done a prior study of NY-ESO-1–directed T cells and saw some evidence of improved response and low toxicity ,” said the trial’s leader, Edward Stadtmauer, M.D., of the University of Pennsylvania. He and his colleagues wanted to see if removing the three genes with CRISPR would make the T cells work even better, he said.
The goal of this study was to first find out if the CRISPR-made treatment was safe. It was tested in two patients with advanced multiple myeloma and one with metastatic sarcoma . All three had tumors that contained NY-ESO-1, the target of the T-cell therapy.
Initial findings suggest that the treatment is safe . Some side effects did occur, but they were likely caused by the chemotherapy patients received before the infusion of NYCE cells, the researchers reported. There was no evidence of an immune reaction to the CRISPR-edited cells.
Only about 10% of the T cells used for the therapy had all four of the desired genetic edits. And off-target edits were found in the modified cells of all three patients. However, none of the cells with off-target edits grew in a way that suggested they had become cancer, Dr. Stadtmauer noted.
The treatment had a small effect on the patients’ cancers. The tumors of two patients (one with multiple myeloma and one with sarcoma) stopped growing for a while but resumed growing later. The treatment didn't work at all for the third patient.
It's exciting that the treatment initially worked for the sarcoma patient because “ solid tumors have been a much more difficult nut to crack with cellular therapy," Dr. Stadtmauer said. "Perhaps [CRISPR] techniques will enhance our ability to treat solid tumors with cell therapies.”
Although the trial shows that CRISPR-edited cell therapy is possible, the long-term effects still need to be monitored, Dr. Stadtmauer continued. The NYCE cells are “safe for as long as we’ve been watching [the study participants]. Our plan is to keep monitoring them for years, if not decades,” he said.
More Studies of CRISPR Treatments to Come
While the study of NYCE T cells marked the first trial of a CRISPR-based cancer treatment, there are likely more to come.
“This [trial] was really a proof-of-principle, feasibility, and safety thing that now opens up the whole world of CRISPR editing and other techniques of [gene] editing to hopefully make the next generation of therapies,” Dr. Stadtmauer said.
Other clinical studies of CRISPR-made cancer treatments are already underway. A few trials are testing CRISPR-engineered CAR T-cell therapies , another type of immunotherapy. For example, one company is testing CRISPR-engineered CAR T cells in people with B cell cancers and people with multiple myeloma .
There are still a lot of questions about all the ways that CRISPR might be put to use in cancer research and treatment. But one thing is for certain: The field is moving incredibly fast and new applications of the technology are constantly popping up.
“People are still improving CRISPR methods,” Dr. Li said. “It’s quite an active area of research and development. I’m sure that CRISPR will have even broader applications in the future.”
Featured Posts
June 5, 2024, by Linda Wang
May 3, 2024, by Carmen Phillips
May 1, 2024, by Edward Winstead
- Biology of Cancer
- Cancer Risk
- Childhood Cancer
- Clinical Trial Results
- Disparities
- FDA Approvals
- Global Health
- Leadership & Expert Views
- Screening & Early Detection
- Survivorship & Supportive Care
- September (1)
- February (6)
- January (6)
- December (7)
- November (6)
- October (7)
- September (7)
- February (7)
- November (7)
- October (5)
- September (6)
- November (4)
- September (9)
- February (5)
- October (8)
- January (7)
- December (6)
- September (8)
- February (9)
- December (9)
- November (9)
- October (9)
- September (11)
- February (11)
- January (10)

Alonzo Mourning, Prostate Cancer Survivor
Online Help
Our 24/7 cancer helpline provides information and answers for people dealing with cancer. We can connect you with trained cancer information specialists who will answer questions about a cancer diagnosis and provide guidance and a compassionate ear.
Chat live online
Select the Live Chat button at the bottom of the page
Call us at 1-800-227-2345
Available any time of day or night
Our highly trained specialists are available 24/7 via phone and on weekdays can assist through online chat. We connect patients, caregivers, and family members with essential services and resources at every step of their cancer journey. Ask us how you can get involved and support the fight against cancer. Some of the topics we can assist with include:
- Referrals to patient-related programs or resources
- Donations, website, or event-related assistance
- Tobacco-related topics
- Volunteer opportunities
- Cancer Information
For medical questions, we encourage you to review our information with your doctor.
Our Research Programs
What does it take to outsmart cancer? Research.
The American Cancer Society (ACS) has helped make possible almost every major cancer breakthrough since 1946. Since then, we've invested more than $5 billion in cancer research, making us the largest nonprofit funder of cancer research in the United States, outside of the federal government.
We remain committed to finding more – and better – ways to improve the quality of life for cancer patients.
Save the Date: June 25-27, 2025 Cancer Research Prevention Conference in London
Investing in Research Pays Off
See how. Check out easy-to-understand summaries of recent studies.

Featured Research Studies

Prostate Cancer Research Highlights
Get the latest research highlights from our prostate cancer research conducted and funded through ACS grants.

Childhood Cancer Research Highlights
The American Cancer Society is committed to finding new answers to help every child and family affected by cancer--see some of our latest research.
We're Finding Answers That Save Lives
Whether we're conducting research or funding it, our goal remains the same: to free the world from the pain and suffering of cancer.

All Cancer Facts & Figures Reports
Learn 2023 estimated new cancer cases and deaths, and stats for breast, colorectal, and global cancer, as well as for African Americans and Hispanics/Latinos.

Cancer Prevention Studies-3 (CPS-3)
See how 300,000 volunteer participants in this active population study are helping us move closer to a world without cancer.

Currently Funded Cancer Research
Learn more about our substantial current spending on research.
Explore Our Research Programs
Our work covers the full realm of cancer research.

Surveillance & Health Equity Science (SHES)
We conduct and publish research on cancer prevention, surveillance, health services, and disparities, including the ACS Cancer Facts & Figures reports.

Population Science (PopSci)
We conduct and publish research about cancer risk factors and the quality of life of cancer survivors, including the ACS Cancer Prevention Studies, CPS-II, and CPS-3.

Extramural Discovery Science (EDS)
We fund high impact and innovative research for any type of cancer and from bench to bedside by supporting scientists across the United States with research grants.

Early Cancer Detection Science
We oversee the development, review, and update of evidence-based cancer screening guidelines using rigorous international standards.

Center for Diversity in Cancer Research (DICR) Training
We fund hands-on research programs for some of today's minority and disadvantaged students to improve the diversity of cancer research and care in the future.
Glossary for Nonscientists
Featured term: cancer survivor.
Anyone who has been diagnosed with cancer, regardless of whether they are actively receiving treatment.
Our Scientists are Helping to End Cancer

Interactive and Multimedia Platforms

Cancer Statistics Center

Cancer Atlas
ACS Research Podcasts
ACS Recorded Webinars
Help us end cancer as we know it, for everyone..
How We Study

Study of Excess Deaths Reveals Racial and Ethnic Disparities
Findings from a large surveillance study led by Meredith Shiels and colleagues published in October 2021 in Annals of Internal Medicine.

Genomic Studies
Investigations of the biological basis of inherited and acquired genetic variants associated with cancer susceptibility.
DCEG investigators utilize a variety of research approaches in seeking to understand the causes of cancer.
- Absolute Risk Modeling
Descriptive Epidemiology
Exposure assessment studies and methods, metabolomics, the human microbiome and cancer, molecular epidemiology, interdisciplinary working groups, psychosocial effects of cancer predisposition syndromes, tumor profiling in relation to cancer etiology, absolute risk modeling.
DCEG investigators in the Biostatistics Branch have developed models (such as the Gail model) for projecting the individualized absolute risk of certain types of cancer. These models have been used to counsel individual patients on their disease risk; to make more formal management recommendations, such as whether or not to take tamoxifen to prevent breast cancer; to design cancer prevention trials; and to assess the potential reductions in population absolute risk from preventive activities. Learn about DCEG absolute risk models for breast cancer, colon cancer, and melanoma.
The Division maintains a broad-ranging, multi-faceted program of descriptive epidemiological studies utilizing a variety of methodological approaches to identify novel risk factors, evaluate tumor heterogeneity, describe current and future trends of common and rare malignancies, and project risk for second primary cancers . See examples of descriptive epidemiology studies .
Evaluating exposure-response relationships is a crucial component in determining cancer causation. For that reason, quantitative exposure assessment plays an essential role in high-quality epidemiologic investigations. DCEG investigators have developed cutting-edge tools and techniques to evaluate the reliability and validity of exposure measurements used in cohort and case-control studies of occupational, lifestyle, and environmental exposures. In addition, our investigators continually work to improve upon these well-established methods. See examples of exposure assessment studies and methods .
DCEG investigates the biological basis of inherited and acquired genetic variants associated with cancer susceptibility, utilizing genome-wide association studies, exome sequencing, and candidate gene studies. DCEG investigators and their collaborators employ an array of advanced statistical methods to support these studies. Learn more about genomic studies .
Metabolomics is the study of small-molecule metabolites in cells, tissues, and organisms that are present in biofluids such as plasma and urine. An emerging field of study, metabolomics has the potential to improve exposure measures and delineate mechanistic links between exposures and cancer. Learn about early-stage DCEG research in metabolomics .
The human microbiota is the collection of all the microorganisms and bacteria that live in or on the human body, such as those present in the digestive system. In the emerging field of microbiomics, researchers study the extent and patterns of these microbes at various body sites and their influence on human health and disease. DCEG investigators are at the forefront of both methodologic and cancer association studies of the microbiome in human populations. Learn about early-stage DCEG research in microbiomics .
We use molecular epidemiology to examine the relationship of genetic and environmental risk factors to cancer etiology. Using laboratory techniques, investigators look for biomarkers of disease and use them to understand the underlying mechanisms of disease in populations. Read about Biomarker Tools developed by the Biostatistics Branch and Clinical Epidemiology Unit .
DCEG scientists form collaborative working groups to enhance the exchange of information and support interdisciplinary approaches to epidemiological and genetic research. Working groups draw their members from across the Division and other organizations in NCI, enabling them to apply a wide range of expertise to the study of complex questions. Read more about Interdisciplinary Working Groups .
The Clinical Genetics Branch investigates and defines best practices of medical, psychosocial, and genetic counseling, as well as risk assessment and communication, to counsel and care for at-risk individuals and families. Read more about research on the Psychosocial Effects of Cancer Predisposition Syndromes .
The recognition of cancer as a heterogeneous disease, coupled with technological advances in molecular/genomic profiling of tumors, provides epidemiologists with the opportunity to integrate tissue profiling in etiologic studies to study the carcinogenesis process, and pinpoint specific factors associated with risk for developing specific molecular or genomic cancer subtypes. DCEG investigators are also undertaking foundational research to identify novel molecular and genomic signatures in tumors linked to germline genetic variants and certain environmental exposures. Learn about tumor profiling research .
Advertisement
Next-Generation Therapeutic Antibodies for Cancer Treatment: Advancements, Applications, and Challenges
- Review Paper
- Published: 02 September 2024
Cite this article

- Abhavya Raja ORCID: orcid.org/0009-0009-7585-4987 1 ,
- Abhishek Kasana ORCID: orcid.org/0000-0002-7403-6490 1 &
- Vaishali Verma ORCID: orcid.org/0000-0001-5076-7435 1
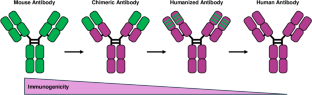
The field of cancer treatment has evolved significantly over the last decade with the emergence of next-generation therapeutic antibodies. Conventional treatments like chemotherapy pose significant challenges, including adverse side effects. Monoclonal antibodies have paved the way for more targeted and effective interventions. The evolution from chimeric to humanized and fully human antibodies has led to a reduction in immunogenicity and enhanced tolerance in vivo. The advent of next-generation antibodies, including bispecific antibodies, nanobodies, antibody-drug conjugates, glyco-engineered antibodies, and antibody fragments, represents a leap forward in cancer therapy. These innovations offer increased potency, adaptability, and reduced drug resistance. Challenges such as target validation, immunogenicity, and high production costs exist. However, technological advancements in antibody engineering techniques provide optimism for addressing these issues. The future promises a paradigm shift, where ongoing research will propel these powerful antibodies to the forefront, revolutionizing the fight against cancer and creating new preventive and curative treatments. This review provides an overview of three next-generation antibody-based molecules, namely bispecific antibodies, antibody-drug conjugates, and nanobodies that have shown promising results in cancer treatment. It discusses the evolution of antibodies from conventional forms to next-generation molecules, along with their applications in cancer treatment, production methods, and associated challenges. The review aims to offer researchers insights into the evolving landscape of next-generation antibody-based cancer therapeutics and their potential to revolutionize treatment strategies.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Subscribe and save.
- Get 10 units per month
- Download Article/Chapter or eBook
- 1 Unit = 1 Article or 1 Chapter
- Cancel anytime
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

Bispecific and multispecific antibodies in oncology: opportunities and challenges

Next generation antibody drugs: pursuit of the 'high-hanging fruit'

Antibody-drug conjugates in cancer therapy: innovations, challenges, and future directions
Sebastian, R., & Nehra, R. (2024). Harnessing the power of adaptive immune response and crosstalk. International Journal of Advanced Biochemistry Research, 8 (1), 06–09. https://doi.org/10.33545/26174693.2024.v8.i1a.276
Article Google Scholar
Oreste, U., Ametrano, A., & Coscia, M. R. (2021). On origin and evolution of the antibody molecule. Biology, 10 (2), 140. https://doi.org/10.3390/biology10020140
Article CAS PubMed PubMed Central Google Scholar
Esposito, S., Amirthalingam, G., Bassetti, M., Blasi, F., De Rosa, F. G., Halasa, N. B., & Principi, N. (2023). Monoclonal antibodies for prophylaxis and therapy of respiratory syncytial virus, SARS-CoV-2, human immunodeficiency virus, rabies and bacterial infections: An update from the world association of infectious diseases and immunological disorders and the Italian society of antinfective therapy. Frontiers in Immunology, 14 , 1162342. https://doi.org/10.3389/fimmu.2023.1162342
Sharma, P., Joshi, R. V., ;, Pritchard, R., ;, Xu, K., ;, Eicher, M. A., Xu, Y., & Eicher, M. A. (2023). Therapeutic antibodies in Medicine. Molecules, Vol. 28 (18), 6438. https://doi.org/10.3390/MOLECULES28186438
Verma, V. (2023). Leveraging monoclonal antibodies as therapeutics to address antimicrobial resistance in bacteria. Journal of Applied Biology & Biotechnology , 11 (3), 53–60. https://doi.org/10.7324/JABB.2023.90087
Article CAS Google Scholar
Wang, Z., Wang, G., Lu, H., Li, H., Tang, M., & Tong, A. (2022). Development of therapeutic antibodies for the treatment of diseases. Molecular biomedicine . https://doi.org/10.1186/S43556-022-00100-4
Article PubMed PubMed Central Google Scholar
Brown, J. S., Amend, S. R., Austin, R. H., Gatenby, R. A., Hammarlund, E. U., & Pienta, K. J. (2023). Updating the definition of cancer. Molecular Cancer Research: MCR, 21 (11), 1142–1147. https://doi.org/10.1158/1541-7786.MCR-23-0411
Article CAS PubMed Google Scholar
Debela, D. T., Muzazu, S. G. Y., Heraro, K. D., Ndalama, M. T., Mesele, B. W., Haile, D. C., Kitui, S. K., & Manyazewal, T. (2021). New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Medicine . https://doi.org/10.1177/20503121211034366
Anand, U., Dey, A., Chandel, A. K., Sanyal, R., Mishra, A., Pandey, D. K., De Falco, V., Upadhyay, A., Kandimalla, R., Chaudhary, A., & Dhanjal, J. K. (2023). Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes & Diseases, 10 (4), 1367–1401. https://doi.org/10.1016/j.gendis.2022.02.007
Paul, S., Konig, M. F., Pardoll, D. M., Bettegowda, C., Papadopoulos, N., Wright, K. M., Gabelli, S. B., Ho, M., van Elsas, A., & Zhou, S. (2024). Cancer therapy with antibodies. Nature Reviews Cancer, 24 (6), 399–426. https://doi.org/10.1038/s41568-024-00690-x
Kumar, M., Jalota, A., Sahu, S. K., & Haque, S. (2024). Therapeutic antibodies for the prevention and treatment of cancer. Journal of Biomedical Science , 31 (1), 6. https://doi.org/10.1186/s12929-024-00996-w
Mitra, S., & Tomar, P. C. (2021). Hybridoma technology; advancements, clinical significance, and future aspects. Journal Genetic Engineering & Biotechnology, 19 (1), 159. https://doi.org/10.1186/s43141-021-00264-6
Pedrioli, A., & Oxenius, A. (2021). Single B cell technologies for monoclonal antibody discovery. Trends in Immunology, 42 (12), 1143–1158. https://doi.org/10.1016/j.it.2021.10.008
Mullard, A. (2021). FDA approves 100th monoclonal antibody product. Nature Reviews Drug Discovery, 20 (7), 491–495. https://doi.org/10.1038/d41573-021-00079-7
Dai, J. M., Zhang, X. Q., Dai, J. Y., Yang, X. M., & Chen, Z. N. (2021). Modified therapeutic antibodies: Improving efficacy. Engineering, 7 (11), 1529–1540. https://doi.org/10.1016/J.ENG.2020.06.030
Verhaar, E. R., Woodham, A. W., & Ploegh, H. L. (2021). Nanobodies in cancer. Seminars in Immunology, 52 , 101425. https://doi.org/10.1016/j.smim.2020.101425
Sedykh, S., Prinz, V., Buneva, V., & Nevinsky, G. (2018). Bispecific antibodies: Design, therapy, perspectives. Drug Design Development and Therapy, 12 , 195–208. https://doi.org/10.2147/DDDT.S151282
Zhang, X., Yang, Y., Fan, D., & Xiong, D. (2017). The development of bispecific antibodies and their applications in tumor immune escape. Experimental Hematology & Oncology . https://doi.org/10.1186/S40164-017-0072-7
Madsen, A. V., Pedersen, L. E., Kristensen, P., & Goletz, S. (2024). Design and engineering of bispecific antibodies: Insights and practical considerations. Frontiers in Bioengineering and Biotechnology, 12 , 1352014. https://doi.org/10.3389/fbioe.2024.1352014
Gu, Y., Wang, Z., & Wang, Y. (2024). Bispecific antibody drug conjugates: Making 1 + 1 > 2. Acta Pharmaceutica Sinica B . https://doi.org/10.1016/J.APSB.2024.01.009
Khongorzul, P., Ling, C. J., Khan, F. U., Ihsan, A. U., & Zhang, J. (2020). Antibody-drug conjugates: A comprehensive review. Molecular Cancer Research, 18 (1), 3–19. https://doi.org/10.1158/1541-7786.MCR-19-0582/82267/
Drago, J. Z., Modi, S., & Chandarlapaty, S. (2021). Unlocking the potential of antibody–drug conjugates for cancer therapy. Nature Reviews Clinical Oncology, 18 (6), 327. https://doi.org/10.1038/S41571-021-00470-8
Su, Z., Xiao, D., Xie, F., Liu, L., Wang, Y., Fan, S., Zhou, X., & Li, S. (2021). Antibody-drug conjugates: Recent advances in linker chemistry. Acta Pharmaceutica Sinica. B, 11 (12), 3889–3907. https://doi.org/10.1016/j.apsb.2021.03.042
Hamers-Casterman, C. T., Atarhouch, T., Muyldermans, S. A., Robinson, G., Hammers, C., Songa, E. B., Bendahman, N., & Hammers, R. (1993). Naturally occurring antibodies devoid of light chains. Nature, 363 (6428), 446–448. https://doi.org/10.1038/363446a0
Jin, B. K., Odongo, S., Radwanska, M., & Magez, S. (2023). Nanobodies: A review of generation, diagnostics and therapeutics. International Journal of Molecular Sciences . https://doi.org/10.3390/IJMS24065994
Sánchez-García, L., Voltà-Durán, E., Parladé, E., Mazzega, E., Sánchez-Chardi, A., Serna, N., López-Laguna, H., Mitstorfer, M., Unzueta, U., Vázquez, E., & Villaverde, A. (2021). Self-assembled nanobodies as selectively targeted, nanostructured, and multivalent materials. ACS Applied Materials & Interfaces, 13 (25), 29406–29415. https://doi.org/10.1021/acsami.1c08092
Kakasi, B., Gácsi, E., Jankovics, H., & Vonderviszt, F. (2023). Extreme thermal stability of the antiGFP nanobody - GFP complex. BMC Research Notes, 16 (1), 110. https://doi.org/10.1186/s13104-023-06382-3
Mei, Y., Chen, Y., Sivaccumar, J. P., An, Z., Xia, N., & Luo, W. (2022). Research progress and applications of nanobody in human infectious diseases. Frontiers in Pharmacology, 13 , 963978. https://doi.org/10.3389/fphar.2022.963978
Kunz, S., Durandy, M., Seguin, L., & Feral, C. C. (2023). NANOBODY ® molecule, a giga medical tool in nanodimensions. International Journal of Molecular Sciences, 24 (17), 13229. https://doi.org/10.3390/IJMS241713229/S1
Babamohamadi, M., Mohammadi, N., Faryadi, E., Haddadi, M., Merati, A., Ghobadinezhad, F., Amirian, R., Izadi, Z., & Hadjati, J. (2024). Anti-CTLA-4 nanobody as a promising approach in cancer immunotherapy. Cell Death & Disease, 15 (1), 17. https://doi.org/10.1038/s41419-023-06391-x
Klein, C., Brinkmann, U., Reichert, J. M., & Kontermann, R. E. (2024). The present and future of bispecific antibodies for cancer therapy. Nature Reviews Drug Discovery, 23 (4), 301–319. https://doi.org/10.1038/s41573-024-00896-6
Ma, J., Mo, Y., Tang, M., Shen, J., Qi, Y., Zhao, W., Huang, Y., Xu, Y., & Qian, C. (2021). Bispecific antibodies: From research to clinical application. Frontiers in immunology . https://doi.org/10.3389/FIMMU.2021.626616
Brinkmann, U., & Kontermann, R. E. (2017). The making of bispecific antibodies. mAbs, 9 (2), 182. https://doi.org/10.1080/19420862.2016.1268307
Lobner, E., Traxlmayr, M. W., Obinger, C., & Hasenhindl, C. (2016). Engineered IgG1-Fc–one fragment to bind them all. Immunological Reviews, 270 (1), 113–131. https://doi.org/10.1111/imr.12385
Mocquot, P., Mossazadeh, Y., Lapierre, L., Pineau, F., & Despas, F. (2022). The pharmacology of blinatumomab: State of the art on pharmacodynamics, pharmacokinetics, adverse drug reactions and evaluation in clinical trials. Journal of Clinical Pharmacy and Therapeutics, 47 (9), 1337–1351. https://doi.org/10.1111/jcpt.13741
Wang, M., Ying, T., & Wu, Y. (2024). Single-domain antibodies as therapeutics for solid tumor treatment. Acta Pharmaceutica Sinica B, 14 (7), 2854–2868. https://doi.org/10.1016/j.apsb.2024.03.016
Yuan, Q., Liang, Q., Sun, Z., Yuan, X., Hou, W., Wang, Y., Wang, H., & Yu, M. (2021). Development of bispecific anti-c-Met/PD-1 diabodies for the treatment of solid tumors and the effect of c-Met binding affinity on efficacy. OncoImmunology, 10 (1), 1914954. https://doi.org/10.1080/2162402X.2021.1914954
Sun, Y., Yu, X., Wang, X., Yuan, K., Wang, G., Hu, L., Zhang, G., Pei, W., Wang, L., Sun, C., & Yang, P. (2023). Bispecific antibodies in cancer therapy: Target selection and regulatory requirements. Acta Pharmaceutica Sinica. B, 13 (9), 3583. https://doi.org/10.1016/J.APSB.2023.05.023
Kontermann, R. E. (2012). Dual targeting strategies with bispecific antibodies. mAbs, 4 (2), 182–197. https://doi.org/10.4161/mabs.4.2.19000
Singh, A., Dees, S., & Grewal, I. S. (2021). Overcoming the challenges associated with CD3 + T-cell redirection in cancer. British Journal of Cancer, 124 (6), 1037–1048. https://doi.org/10.1038/s41416-020-01225-5
Yang, Y., Wu, H., Yang, Y., Kang, Y., He, R., Zhou, B., Guo, H., Zhang, J., Li, J., Ge, C., & Wang, T. (2023). Dual blockade of CD47 and CD24 signaling using a novel bispecific antibody fusion protein enhances macrophage immunotherapy. Molecular Therapy - Oncolytics, 31 , 100747
Google Scholar
Nikkhoi, S. K., Li, G., Eleya, S., Yang, G., Vandavasi, V. G., & Hatefi, A. (2023). Bispecific killer cell engager with high affinity and specificity toward CD16a on NK cells for cancer immunotherapy. Frontiers in Immunology, 13 , 1039969. https://doi.org/10.3389/fimmu.2022.1039969
Liu, X., Zhao, J., Guo, X., & Song, Y. (2023). CD20 × CD3 bispecific antibodies for lymphoma therapy: Latest updates from ASCO 2023 annual meeting. Journal of Hematology & Oncology, 16 (1), 90. https://doi.org/10.1186/s13045-023-01488-4
Huo, J., Huang, Y., Zheng, Z., Tay, X. N., Mahfut, F. B., Zhang, W., Lam, K. P., Yang, Y., & Xu, S. (2022). Development of a T cell-redirecting bispecific antibody targeting B-cell maturation antigen for the suppression of multiple myeloma cell growth. Antibody Therapeutics, 5 (2), 138–149. https://doi.org/10.1093/abt/tbac012
Rodriguez-Otero, P., van de Donk, N. W., Pillarisetti, K., Cornax, I., Vishwamitra, D., Gray, K., Hilder, B., Tolbert, J., Renaud, T., Masterson, T., & Heuck, C. (2024). GPRC5D as a novel target for the treatment of multiple myeloma: A narrative review. Blood Cancer Journal, 14 (1), 24. https://doi.org/10.1038/s41408-023-00966-9
Martinez-Perez, D., Viñal, D., Solares, I., Espinosa, E., & Feliu, J. (2021). Gp-100 as a novel therapeutic target in uveal melanoma. Cancers, 13 (23), 5968. https://doi.org/10.3390/cancers13235968
Majumder, A. (2023). HER3: Toward the prognostic significance, therapeutic potential, current challenges, and future therapeutics in different types of cancer. Cells, 12 (21), 2517. https://doi.org/10.3390/cells12212517
Fontana, E., Torga, G., Fostea, R., Cleator, S., Wasserman, E., Murat, A., & Arkenau, H. T. (2022). Sustained tumor regression with zenocutuzumab, a bispecific antibody targeting human epidermal growth factor receptor 2/human epidermal growth factor receptor 3 signaling, in NRG1 Fusion-positive, estrogen receptor-positive breast cancer after progression on a cyclin-dependent kinase 4/6 inhibitor. JCO Precision Oncology, 6 , e2100446. https://doi.org/10.1200/PO.21.00446
Article PubMed Google Scholar
Cho, B. C., Simi, A., Sabari, J., Vijayaraghavan, S., Moores, S., & Spira, A. (2023). Amivantamab, an epidermal growth factor receptor (EGFR) and mesenchymal-epithelial transition factor (MET) bispecific antibody, designed to enable multiple mechanisms of action and broad clinical applications. Clinical Lung Cancer, 24 (2), 89–97. https://doi.org/10.1016/j.cllc.2022.11.004
You, W. K., Schuetz, T. J., & Lee, S. H. (2023). Targeting the DLL/Notch signaling pathway in cancer: Challenges and advances in clinical development. Molecular Cancer Therapeutics, 22 (1), 3–11. https://doi.org/10.1158/1535-7163.MCT-22-0243
Liu, N., Liu, M., Fu, S., Wang, J., Tang, H., Isah, A. D., Chen, D., & Wang, X. (2022). Ang2-targeted combination therapy for cancer treatment. Frontiers in Immunology, 13 , 949553
Zarrabi, K. K., Narayan, V., Mille, P. J., Zibelman, M. R., Miron, B., Bashir, B., & Kelly, W. K. (2023). Bispecific PSMA antibodies and CAR-T in metastatic castration-resistant prostate cancer. Therapeutic Advances in Urology, 15 , 17562872231182220. https://doi.org/10.1177/17562872231182219
Zhang, T., Lin, Y., & Gao, Q. (2023). Bispecific antibodies targeting immunomodulatory checkpoints for cancer therapy. Cancer Biology & Medicine, 20 (3), 181–195. https://doi.org/10.20892/j.issn.2095-3941.2023.0002
Cheng, L., Chen, L., Shi, Y., Gu, W., Ding, W., Zheng, X., Liu, Y., Jiang, J., & Zheng, Z. (2024). Efficacy and safety of bispecific antibodies vs. immune checkpoint blockade combination therapy in cancer: A real-world comparison. Molecular Cancer, 23 (1), 77. https://doi.org/10.1186/s12943-024-01956-6
The Antibody Society. (2024). Therapeutic monoclonal antibodies approved or in regulatory review. Retrieved August 16, 2024. https://doi.org/www.antibodysociety.org/antibody-therapeutics-product-data
Burt, R., Warcel, D., & Fielding, A. K. (2019). Blinatumomab, a bispecific B-cell and T-cell engaging antibody, in the treatment of B-cell malignancies. Human Vaccines & Immunotherapeutics, 15 (3), 594–602. https://doi.org/10.1080/21645515.2018.1540828
Pang, X., Huang, Z., Zhong, T., Zhang, P., Wang, Z. M., Xia, M., & Li, B. (2023). Cadonilimab, a tetravalent PD-1/CTLA-4 bispecific antibody with trans-binding and enhanced target binding avidity. mAbs, 15 (1), 2180794. https://doi.org/10.1080/19420862.2023.2180794
Wang, S., Chen, K., Lei, Q., Ma, P., Yuan, A. Q., Zhao, Y., Jiang, Y., Fang, H., Xing, S., Fang, Y., & Jiang, N. (2021). The state of the art of bispecific antibodies for treating human malignancies. EMBO Molecular Medicine, 13 (9), e14291
Wang, Z., Li, H., Gou, L., Li, W., & Wang, Y. (2023). Antibody–drug conjugates: Recent advances in payloads. Acta Pharmaceutica Sinica B, 13 (10), 4025. https://doi.org/10.1016/J.APSB.2023.06.015
Fu, Z., Li, S., Han, S., Shi, C., & Zhang, Y. (2022). Antibody drug conjugate: The biological missile for targeted cancer therapy. Signal Transduction and Targeted Therapy, 2022 7:1 (1), 1–25. https://doi.org/10.1038/s41392-022-00947-7
Kuwatani, M., & Sakamoto, N. (2023). Promising highly targeted therapies for cholangiocarcinoma: A review and future perspectives. Cancers . https://doi.org/10.3390/CANCERS15143686
Riccardi, F., Bo, M. D., Macor, P., & Toffoli, G. (2023). A comprehensive overview on antibody-drug conjugates: from the conceptualization to cancer therapy. Frontiers in Pharmacology . https://doi.org/10.3389/FPHAR.2023.1274088/FULL
Song, C. H., Jeong, M., In, H., Kim, J. H., Lin, C. W., & Han, K. H. (2023). Trends in the development of antibody-drug conjugates for cancer therapy. Antibodies, 12 (4), 72. https://doi.org/10.3390/ANTIB12040072/S1
Strop, P., Delaria, K., Foletti, D., Witt, J. M., Hasa-Moreno, A., Poulsen, K., Casas, M. G., Dorywalska, M., Farias, S., Pios, A., & Lui, V. (2015). Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nature Biotechnology, 33 (7), 694–696. https://doi.org/10.1038/nbt.3274
Zacharias, N., Podust, V. N., Kajihara, K. K., Leipold, D., Del Rosario, G., Thayer, D., Dong, E., Paluch, M., Fischer, D., Zheng, K., & Lei, C. (2022). A homogeneous high-DAR antibody–drug conjugate platform combining THIOMAB antibodies and XTEN polypeptides. Chemical Science, 13 (11), 3147–3160. https://doi.org/10.1039/D1SC05243H
Jäger, S., Wagner, T. R., Rasche, N., Kolmar, H., Hecht, S., & Schröter, C. (2021). Generation and biological evaluation of fc antigen binding fragment-drug conjugates as a novel antibody-based format for targeted drug delivery. Bioconjugate Chemistry, 32 (8), 1699–1710. https://doi.org/10.1021/acs.bioconjchem.1c00240
Simmons, J. K., Burke, P. J., Cochran, J. H., Pittman, P. G., & Lyon, R. P. (2020). Reducing the antigen-independent toxicity of antibody-drug conjugates by minimizing their non-specific clearance through PEGylation. Toxicology and Applied Pharmacology, 392 , 114932. https://doi.org/10.1016/j.taap.2020.114932
Dai, L. J., Li, Y. W., Ma, D., Shao, Z. M., & Jiang, Y. Z. (2023). Next-generation antibody–drug conjugates revolutionize the precise classification and treatment of HER2-expressing breast cancer. Cancer Biology & Medicine, 20 (10), 689. https://doi.org/10.20892/J.ISSN.2095-3941.2023.0286
de Goeij, B. E., Vink, T., Ten Napel, H., Breij, E. C., Satijn, D., Wubbolts, R., Miao, D., & Parren, P. W. (2016). Efficient payload delivery by a bispecific antibody-drug conjugate targeting HER2 and CD63. Molecular Cancer Therapeutics, 15 (11), 2688–2697. https://doi.org/10.1158/1535-7163.MCT-16-0364
Schoenfeld, K., Harwardt, J., Habermann, J., Elter, A., & Kolmar, H. (2023). Conditional activation of an anti-IgM antibody-drug conjugate for precise B cell lymphoma targeting. Frontiers in Immunology, 14 , 1258700. https://doi.org/10.3389/fimmu.2023.1258700
Gerber, H. P., Sapra, P., Loganzo, F., & May, C. (2016). Combining antibody–drug conjugates and immune-mediated cancer therapy: What to expect? Biochemical Pharmacology, 102 , 1–6. https://doi.org/10.1016/j.bcp.2015.12.008
Sheyi, R., de Torre, B. G., & Albericio, F. (2022). Linkers: An assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics . https://doi.org/10.3390/PHARMACEUTICS14020396
Greenberg, A. S., Avila, D., Hughes, M., Hughes, A., McKinney, E. C., & Flajnik, M. F. (1995). A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature, 374 (6518), 168–173. https://doi.org/10.1038/374168a0
Rizk, S. S., Moustafa, D. M., ElBanna, S. A., El-Din, N., & Attia, A. S. (2024). Nanobodies in the fight against infectious diseases: Repurposing nature’s tiny weapons. World Journal of Microbiology and Biotechnology, 40 (7), 209. https://doi.org/10.1007/s11274-024-03990-4
Santos, L., Moreira, J. N., Abrunhosa, A., & Gomes, C. (2024). Brain metastasis: An insight into novel molecular targets for theranostic approaches. Critical Reviews in Oncology/Hematology, 198 , 104377. https://doi.org/10.1016/j.critrevonc.2024.104377
Jumapili, N. A., Zivalj, M., Barthelmess, R. M., Raes, G., De Groof, T. W., Devoogdt, N., Stijlemans, B., Vincke, C., & Van Ginderachter, J. A. (2023). A few good reasons to use nanobodies for cancer treatment. European Journal of Immunology, 53 (9), 2250024. https://doi.org/10.1002/eji.202250024
Ji, F., Ren, J., Vincke, C., Jia, L., & Muyldermans, S. (2022). Nanobodies: From serendipitous discovery of heavy chain-only antibodies in camelids to a wide range of useful applications. Methods in Molecular Biology (Clifton N J), 2446 , 3–17. https://doi.org/10.1007/978-1-0716-2075-5_1
Bao, G., Tang, M., Zhao, J., & Zhu, X. (2021). Nanobody: A promising toolkit for molecular imaging and disease therapy. EJNMMI Research, 11 (1), 1–13. https://doi.org/10.1186/s13550-021-00750-5
Yep, A. T., Takeuchi, Y., Engelhardt, O. G., & Hufton, S. E. (2021). Broad reactivity single domain antibodies against influenza virus and their applications to vaccine potency testing and immunotherapy. Biomolecules, 11 (3), 1–23. https://doi.org/10.3390/BIOM11030407
Zhang, Q., Zhang, N., Xiao, H., Wang, C., & He, L. (2023). Small antibodies with big applications: Nanobody-based cancer diagnostics and therapeutics. Cancers . https://doi.org/10.3390/CANCERS15235639
Wesolowski, J., Alzogaray, V., Reyelt, J., Unger, M., Juarez, K., Urrutia, M., Cauerhff, A., Danquah, W., Rissiek, B., Scheuplein, F., & Schwarz, N. (2009). Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Medical Microbiology and Immunology, 198 (3), 157–174. https://doi.org/10.1007/S00430-009-0116-7
Wang, J., Kang, G., Yuan, H., Cao, X., Huang, H., & de Marco, A. (2021). Research progress and applications of multivalent, multispecific and modified nanobodies for disease treatment. Frontiers in Immunology. https://doi.org/10.3389/FIMMU.2021.838082
Shoari, A., Tahmasebi, M., Khodabakhsh, F., Cohan, R. A., Oghalaie, A., & Behdani, M. (2022). Angiogenic biomolecules specific nanobodies application in cancer imaging and therapy; review and updates. International Immunopharmacology, 105 , 108585. https://doi.org/10.1016/j.intimp.2022.108585
Bolli, E., Scherger, M., Arnouk, S. M., Pombo Antunes, A. R., Straßburger, D., Urschbach, M., Stickdorn, J., De Vlaminck, K., Movahedi, K., Räder, H. J., & Hernot, S. (2021). Targeted repolarization of tumor-associated macrophages via imidazoquinoline-linked nanobodies. Advanced Science, 8 (10), 2004574. https://doi.org/10.1002/advs.202004574
Dougan, M., Ingram, J. R., Jeong, H. J., Mosaheb, M. M., Bruck, P. T., Ali, L., Pishesha, N., Blomberg, O., Tyler, P. M., Servos, M. M., & Rashidian, M. (2018). Targeting cytokine therapy to the pancreatic tumor microenvironment using PD-L1–specific VHHs. Cancer Immunology Research, 6 (4), 389–401. https://doi.org/10.1158/2326-6066.CIR-17-0495
de Bruin, R. C., Veluchamy, J. P., Lougheed, S. M., Schneiders, F. L., Lopez-Lastra, S., Lameris, R., Stam, A. G., Sebestyen, Z., Kuball, J., Molthoff, C. F., & Hooijberg, E. (2018). A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vδ2-T cells. OncoImmunology, 7 (1), e1375641
Boutin, L., Barjon, C., Chauvet, M., Lafrance, L., Senechal, E., Bourges, D., Vigne, E., & Scotet, E. (2024). Camelid-derived Tcell engagers harnessing human γδ T cells as promising antitumor immunotherapeutic agents. European Journal of Immunology, 54 (8), 2350773. https://doi.org/10.1002/eji.202350773
Safarzadeh Kozani, P., Naseri, A., Mirarefin, S. M. J., Salem, F., Nikbakht, M., Bakhshi, E., & Kozani, S. (2022). Nanobody-based CAR-T cells for cancer immunotherapy. Biomarker Research, 10 (1), 24. https://doi.org/10.1186/s40364-022-00371-7
Xia, B., Lin, K., Wang, X., Chen, F., Zhou, M., Li, Y., Lin, Y., Qiao, Y., Li, R., Zhang, W., & He, X. (2023). Nanobody-derived bispecific CAR-T cell therapy enhances the anti-tumor efficacy of T cell lymphoma treatment. Molecular Therapy - Oncolytics, 30 , 86–102. https://doi.org/10.1016/j.omto.2023.07.007
Jin, S., Sun, Y., Liang, X., Gu, X., Ning, J., Xu, Y., Chen, S., & Pan, L. (2022). Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduction and Targeted Therapy . https://doi.org/10.1038/S41392-021-00868-X
De Pauw, T., De Mey, L., Debacker, J. M., Raes, G., Van Ginderachter, J. A., De Groof, T. W. M., & Devoogdt, N. (2023). Current status and future expectations of nanobodies in oncology trials. Expert Opinion on Investigational Drugs, 32 (8), 705–721. https://doi.org/10.1080/13543784.2023.2249814
Davis, J., McGann, M., Shockley, A., & Hashmi, H. (2022). Idecabtagene vicleucel versus ciltacabtagene autoleucel: A Sophie’s choice for patients with relapsed refractory multiple myeloma. Expert Review of Hematology, 15 (6), 473–475. https://doi.org/10.1080/17474086.2022.2081147
Emmerich, C. H., Gamboa, L. M., Hofmann, M. C., Bonin-Andresen, M., Arbach, O., Schendel, P., Gerlach, B., Hempel, K., Bespalov, A., Dirnagl, U., & Parnham, M. J. (2021). Improving target assessment in biomedical research: The GOT-IT recommendations. Nature Reviews. Drug Discovery, 20 (1), 64. https://doi.org/10.1038/S41573-020-0087-3
Pan, D., & Richter, J. (2023). Teclistamab for multiple myeloma: Clinical insights and practical considerations for a first-in-class bispecific antibody. Cancer Management and Research, 15 , 741–751. https://doi.org/10.2147/CMAR.S372237
Esapa, B., Jiang, J., Cheung, A., Chenoweth, A., Thurston, D. E., & Karagiannis, S. N. (2023). Target antigen attributes and their contributions to clinically approved antibody-drug conjugates (ADCs) in haematopoietic and solid cancers. Cancers . https://doi.org/10.3390/CANCERS15061845
Peters, C., & Brown, S. (2015). Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Bioscience Reports, 35 (4), 225. https://doi.org/10.1042/BSR20150089
Valldorf, B., Hinz, S. C., Russo, G., Pekar, L., Mohr, L., Klemm, J., Doerner, A., Krah, S., Hust, M., & Zielonka, S. (2022). Antibody display technologies: Selecting the cream of the crop. Biological Chemistry, 403 (5–6), 455–477. https://doi.org/10.1515/hsz-2020-0377
Zhang, Y. (2023). Evolution of phage display libraries for therapeutic antibody discovery. mAbs . https://doi.org/10.1080/19420862.2023.2213793
Almagro, J. C., Pedraza-Escalona, M., Arrieta, H. I., & Pérez-Tapia, S. M. (2019). Phage display libraries for antibody therapeutic discovery and development. Antibodies . https://doi.org/10.3390/ANTIB8030044
Svilenov, H. L., Arosio, P., Menzen, T., Tessier, P., & Sormanni, P. (2023). Approaches to expand the conventional toolbox for discovery and selection of antibodies with drug-like physicochemical properties. mAbs . https://doi.org/10.1080/19420862.2022.2164459
Sheehan, J., & Marasco, W. A. (2015). Phage and yeast display. Microbiology Spectrum . https://doi.org/10.1128/MICROBIOLSPEC.AID-0028-2014
Kunamneni, A., Ogaugwu, C., Bradfute, S., & Durvasula, R. (2020). Ribosome Display Technology: Applications in Disease. Diagnosis and Control Antibodies, 9 (3), 1–17. https://doi.org/10.3390/ANTIB9030028
Chen, W. C., & Murawsky, C. M. (2018). Strategies for generating diverse antibody repertoires using transgenic animals expressing human antibodies. Frontiers in Immunology, 9 , 460. https://doi.org/10.3389/fimmu.2018.00460
Kim, J., McFee, M., Fang, Q., Abdin, O., & Kim, P. M. (2023). Computational and artificial intelligence-based methods for antibody development. Trends in Pharmacological Sciences, 44 (3), 175–189. https://doi.org/10.1016/J.TIPS.2022.12.005
Joubbi, S., Micheli, A., Milazzo, P., Maccari, G., Ciano, G., Cardamone, D., & Medini, D. (2024). Antibody design using deep learning: From sequence and structure design to affinity maturation. Briefings in Bioinformatics, 25 (4), bbae307. https://doi.org/10.1093/bib/bbae307
Porebski, B. T., Balmforth, M., Browne, G., Riley, A., Jamali, K., Fürst, M. J., Velic, M., Buchanan, A., Minter, R., Vaughan, T., & Holliger, P. (2023). Rapid discovery of high-affinity antibodies via massively parallel sequencing, ribosome display and affinity screening. Nature Biomedical Engineering, 2023 , 1–19. https://doi.org/10.1038/s41551-023-01093-3
Manieri, T. M., Magalhaes, C. G., Takata, D. Y., Batalha-Carvalho, J. V., & Moro, A. M. (2020). In silico techniques for prospecting and characterizing monoclonal antibodies. Monoclonal Antibodies . https://doi.org/10.5772/INTECHOPEN.94366
Hadsund, J. T., Satława, T., Janusz, B., Shan, L., Zhou, L., Röttger, R., & Krawczyk, K. (2024). nanoBERT: A deep learning model for gene agnostic navigation of the nanobody mutational space. Bioinformatics Advances, 4 (1), vbae033. https://doi.org/10.1093/bioadv/vbae033
Wossnig, L., Furtmann, N., Buchanan, A., Kumar, S., & Greiff, V. (2024). Best practices for machine learning in antibody discovery and development. Drug Discovery Today, 29 (7), 104025. https://doi.org/10.1016/j.drudis.2024.104025
Matsunaga, R., Ujiie, K., Inagaki, M., Fernández Pérez, J., Yasuda, Y., Mimasu, S., Soga, S., & Tsumoto, K. (2023). High-throughput analysis system of interaction kinetics for data-driven antibody design. Scientific Reports, 13 (1), 1–9. https://doi.org/10.1038/s41598-023-46756-y
Makowski, E. K., Wu, L., Desai, A. A., & Tessier, P. M. (2021). Highly sensitive detection of antibody nonspecific interactions using flow cytometry. mAbs . https://doi.org/10.1080/19420862.2021.1951426
Francino-Urdaniz, I. M., & Whitehead, T. A. (2021). An overview of methods for the structural and functional mapping of epitopes recognized by anti-SARS-CoV-2 antibodies. RSC Chemical Biology, 2 (6), 1580–1589. https://doi.org/10.1039/d1cb00169h
Jethva, P. N., & Gross, M. L. (2023). Hydrogen deuterium exchange and other mass spectrometry- based approaches for epitope mapping. Frontiers in Analytical Science, 3 , 1118749. https://doi.org/10.3389/FRANS.2023.1118749
Verma, V., Joshi, G., Gupta, A., & Chaudhary, V. K. (2020). An efficient ORF selection system for DNA fragment libraries based on split beta-lactamase complementation. PloS ONE, 15 (7), e0235853.
Jin, P., & Zhu, Z. (2011). The design and engineering of IgG-like bispecific antibodies. Bispecific Antibodies . https://doi.org/10.1007/978-3-642-20910-9_9
Fawcett, C., Tickle, J. R., & Coles, C. H. (2024). Facilitating high throughput bispecific antibody production and potential applications within biopharmaceutical discovery workflows. mAbs . https://doi.org/10.1080/19420862.2024.2311992
Article PubMed Central Google Scholar
Vaur, V., Koutsopetras, I., Erb, S., Jackowska, B., Benazza, R., Cahuzac, H., Detappe, A., Hernandez-Alba, O., Cianférani, S., Scott, C. J., & Chaubet, G. (2024). Chemical production of cytotoxic bispecific antibodies using the UGI multicomponent reaction. ChemBioChem . https://doi.org/10.1002/cbic.202400170
Dimasi, N., Kumar, A., & Gao, C. (2021). Generation of bispecific antibodies using chemical conjugation methods. Drug Discovery Today: Technologies, 40 , 13–24. https://doi.org/10.1016/j.ddtec.2021.08.006
Singh, R., Chandley, P., & Rohatgi, S. (2023). Recent advances in the development of monoclonal antibodies and next-generation antibodies. ImmunoHorizons, 7 (12), 886–897. https://doi.org/10.4049/immunohorizons.2300102
Moon, D., Tae, N., Park, Y., Lee, S. W., & Kim, D. H. (2022). Development of bispecific antibody for Cancer Immunotherapy: Focus on T cell engaging antibody. Immune Network, 22 (1), e4. https://doi.org/10.4110/in.2022.22.e4
Xu, Y., Lee, J., Tran, C., Heibeck, T. H., Wang, W. D., Yang, J., Stafford, R. L., Steiner, A. R., Sato, A. K., Hallam, T. J., & Yin, G. (2015). Production of bispecific antibodies in “knobs-into-holes” using a cell-free expression system. mAbs, 7 (1), 231. https://doi.org/10.4161/19420862.2015.989013
Klein, C., Schaefer, W., & Regula, J. T. (2016). The use of CrossMAb technology for the generation of bi- and multispecific antibodies. mAbs, 8 (6), 1010. https://doi.org/10.1080/19420862.2016.1197457
Fernandez-Martinez, D., Tully, M. D., Leonard, G., Mathieu, M., & Kandiah, E. (2023). Structural insights into the bi-specific cross-over dual variable antibody architecture by cryo-EM. Scientific Reports 2023, 13:1 (1), 1–11. https://doi.org/10.1038/s41598-023-35678-4
Rashid, M. H. (2022). Full-length recombinant antibodies from Escherichia coli : Production, characterization, effector function (fc) engineering, and clinical evaluation. mAbs, 14 (1), 2111748. https://doi.org/10.1080/19420862.2022.2111748
Tripathi, N. K., & Shrivastava, A. (2019). Recent developments in bioprocessing of recombinant proteins: Expression hosts and process development. Frontiers in Bioengineering and Biotechnology, 7 , 420. https://doi.org/10.3389/fbioe.2019.00420
Kostova, V., Désos, P., Starck, J. B., & Kotschy, A. (2021). The chemistry behind ADCs. Pharmaceuticals . https://doi.org/10.3390/PH14050442
Zhou, Q. (2017). Site-specific antibody conjugation for ADC and beyond. Biomedicines . https://doi.org/10.3390/BIOMEDICINES5040064
Dudchak, R., Podolak, M., Holota, S., Szewczyk-Roszczenko, O., Roszczenko, P., Bielawska, A., & Bielawski, K. (2024). Click chemistry in the synthesis of antibody-drug conjugates. Bioorganic Chemistry, 19 ,106982
Su, Q., Shi, W., Huang, X., Yin, S., Yang, X., & Lu, X. (2023). Recent advances of nanobody applications in diagnosis and detection. MedComm – Biomaterials and Applications, 2 (3), e54. https://doi.org/10.1002/MBA2.54
de Marco, A. (2020). Recombinant expression of nanobodies and nanobody-derived immunoreagents. Protein Expression and Purification, 172 , 105645. https://doi.org/10.1016/J.PEP.2020.105645
Nguyen, D. H., Chong, A., Hong, Y., & Min, J. J. (2023). Bioengineering of bacteria for cancer immunotherapy. Nature Communications, 14 (1), 3553. https://doi.org/10.1038/s41467-023-39224-8
Liu, L., Liu, X., Xin, W., Zhou, L., Huang, B., Han, C., Cao, Z., & Hua, Z. (2023). A bacteria-based system expressing anti-TNF-α nanobody for enhanced cancer immunotherapy. Signal Transduction and Targeted Therapy, 8 (1), 134. https://doi.org/10.1038/s41392-023-01364-0
Zhao, X., Rahman, M., Xu, Z., Kasputis, T., He, Y., Yuan, L., Wright, R. C., & Chen, J. (2023). Engineered yeast displaying specific norovirus-binding nanobodies for the concentration and detection of human norovirus in food matrix. Journal of Agricultural and Food Chemistry, 71 (22), 8665–8672. https://doi.org/10.1021/acs.jafc.3c01946
Zheng, Y., Li, B., Zhao, S., Liu, J., & Li, D. (2024). A universal strategy for the efficient expression of nanobodies in Pichia pastoris . Fermentation, 10 (1), 37. https://doi.org/10.3390/fermentation10010037
Wang, Y., Li, X., Chen, X., Nielsen, J., Petranovic, D., & Siewers, V. (2021). Expression of antibody fragments in Saccharomyces cerevisiae strains evolved for enhanced protein secretion. Microbial Cell Factories, 20 (1), 134. https://doi.org/10.1186/s12934-021-01624-0
Hemmer, C., Djennane, S., Ackerer, L., Hleibieh, K., Marmonier, A., Gersch, S., Garcia, S., Vigne, E., Komar, V., Perrin, M., & Gertz, C. (2018). Nanobody-mediated resistance to Grapevine fanleaf virus in plants. Plant Biotechnology Journal, 16 (2), 660–671. https://doi.org/10.1111/pbi.12819
Park, S. R., Lee, J. H., Kim, K., Kim, T. M., Lee, S. H., Choo, Y. K., Kim, K. S., & Ko, K. (2020). Expression and in vitro function of anti-breast cancer llama-based single domain antibody VHH expressed in tobacco plants. International Journal of Molecular Sciences, 21 (4), 1354. https://doi.org/10.3390/ijms21041354
Park, C., Kim, K., Kim, Y., Zhu, R., Hain, L., Seferovic, H., Kim, M. H., Woo, H. J., Hwang, H., Lee, S. H., & Kim, S. (2024). Plant-derived anti-human epidermal growth factor receptor 2 antibody suppresses trastuzumab-resistant breast cancer with enhanced nanoscale binding. ACS Nano, 18 (25), 16126–16140. https://doi.org/10.1021/acsnano.4c00360
Jin, C., Kang, Y. J., Park, S. R., Oh, Y. J., & Ko, K. (2024). Production, expression, and function of dual-specific monoclonal antibodies in a single plant. Planta, 259 (1), 15. https://doi.org/10.1007/s00425-023-04284-z
Smolskaya, S., Logashina, Y. A., & Andreev, Y. A. (2020). Escherichia coli extract-based cell-free expression system as an alternative for difficult-to-obtain protein biosynthesis. International Journal of Molecular Sciences , 21 (3), 928. https://doi.org/10.3390/IJMS21030928
Simão, D. C., Zarrabi, K. K., Mendes, J. L., Luz, R., Garcia, J. A., Kelly, W. K., & Barata, P. C. (2023). Bispecific T-cell engagers therapies in solid tumors: focusing on prostate cancer. Cancers, 15 (5), 1412. https://doi.org/10.3390/cancers15051412
Lesokhin, A. M., Tomasson, M. H., Arnulf, B., Bahlis, N. J., Miles Prince, H., Niesvizky, R., Rodrίguez-Otero, P., Martinez-Lopez, J., Koehne, G., Touzeau, C., & Jethava, Y. (2023). Elranatamab in relapsed or refractory multiple myeloma: Phase 2 magnetisMM-3 trial results. Nature Medicine, 29 (9), 2259–2267. https://doi.org/10.1038/s41591-023-02528-9
Chari, A., Minnema, M. C., Berdeja, J. G., Oriol, A., van de Donk, N. W., Rodríguez-Otero, P., Askari, E., Mateos, M. V., Costa, L. J., Caers, J., & Verona, R. (2022). Talquetamab, a T-cell–redirecting GPRC5D bispecific antibody for multiple myeloma. New England Journal of Medicine, 387 (24), 2232–2244. https://doi.org/10.1056/NEJMoa2204591
Howlett, S., Carter, T. J., Shaw, H. M., & Nathan, P. D. (2023). Tebentafusp: A first-in-class treatment for metastatic uveal melanoma. Therapeutic Advances in Medical Oncology, 15 , 17588359231160140. https://doi.org/10.1177/17588359231160140
Dhillon, S. (2024). Tarlatamab: First approval. Drugs . https://doi.org/10.1007/s40265-024-02070-z
Chon, K., Larkins, E., Chatterjee, S., Mishra-Kalyani, P. S., Aungst, S., Wearne, E., Subramaniam, S., Li, Y., Liu, J., Sun, J., & Charlab, R. (2023). FDA approval summary: Amivantamab for the treatment of patients with non-small cell lung cancer with EGFR exon 20 insertion mutations. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 29 (17), 3262–3266. https://doi.org/10.1158/1078-0432.CCR-22-3713
Brazel, D., & Nagasaka, M. (2023). The development of amivantamab for the treatment of non-small cell lung cancer. Respiratory Research, 24 (1), 256. https://doi.org/10.1186/s12931-023-02558-4
Dhillon, S. (2024). Ivonescimab: First approval. Drugs . https://doi.org/10.1007/s40265-024-02073-w
Mckertish, C. M., & Kayser, V. (2021). Advances and limitations of antibody drug conjugates for cancer. Biomedicines . https://doi.org/10.3390/BIOMEDICINES9080872
Mullard, A. (2022). FDA approves second BCMA-targeted CAR-T cell therapy. Nature Reviews Drug Discovery, 21 (4), 249. https://doi.org/10.1038/d41573-022-00048-8
Hughes, J. P., Rees, S. S., Kalindjian, S. B., & Philpott, K. L. (2011). Principles of early drug discovery. British Journal of Pharmacology, 162 (6), 1239. https://doi.org/10.1111/J.1476-5381.2010.01127.X
Zhang, A., Miao, K., Sun, H., & Deng, C. X. (2022). Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. International Journal of Biological Sciences, 18 (7), 3019. https://doi.org/10.7150/IJBS.72534
Goulet, D. R., & Atkins, W. M. (2020). Considerations for the design of antibody-based therapeutics. Journal of Pharmaceutical Sciences, 109 (1), 74. https://doi.org/10.1016/J.XPHS.2019.05.031
Qin, X., Ning, W., Liu, H., Liu, X., Luo, W., & Xia, N. (2024). Stepping forward: T-cell redirecting bispecific antibodies in cancer therapy. Acta Pharmaceutica Sinica B, 14 (6), 2361–2377. https://doi.org/10.1016/j.apsb.2024.03.027
Tacchetti, P., Barbato, S., Mancuso, K., Zamagni, E., & Cavo, M. (2024). Bispecific antibodies for the management of relapsed/refractory multiple myeloma. Cancers, 16 (13), 2337. https://doi.org/10.3390/cancers16132337
Tsuchikama, K., Anami, Y., Ha, S. Y. Y., & Yamazaki, C. M. (2024). Exploring the next generation of antibody–drug conjugates. Nature Reviews Clinical Oncology, 21 (3), 203–223. https://doi.org/10.1038/s41571-023-00850-2
Segués, A., Huang, S., Sijts, A., Berraondo, P., & Zaiss, D. M. (2022). Opportunities and challenges of bi-specific antibodies. International Review of Cell and Molecular Biology , 369 , 45–70. https://doi.org/10.1016/bs.ircmb.2022.05.001
Wei, J., Yang, Y., Wang, G., & Liu, M. (2022). Current landscape and future directions of bispecific antibodies in cancer immunotherapy. Frontiers in Immunology, 13 , 1035276. https://doi.org/10.3389/fimmu.2022.1035276
Gupta, N., Geethika, L. S., & Sneha, P. (2024). Antibody-drug Conjugates in Cancer Treatment: An overview. Journal of Cancer and Tumor International, 14 (3), 33–45. https://doi.org/10.9734/jcti/2024/v14i3259
Zhang, B., Wang, M., Sun, L., Liu, J., Yin, L., Xia, M., Zhang, L., Liu, X., & Cheng, Y. (2024). Recent advances in targeted cancer therapy: Are PDCs the next generation of ADCs? Journal of Medicinal Chemistry, 67 (14), 11469–11487. https://doi.org/10.1021/acs.jmedchem.4c00106
Jin, Y., Schladetsch, M. A., Huang, X., Balunas, M. J., & Wiemer, A. J. (2022). Stepping forward in antibody-drug conjugate development. Pharmacology & Therapeutics, 229 , 107917. https://doi.org/10.1016/j.pharmthera.2021.107917
Wu, J., Lu, H., Xu, X., Rao, L., & Ge, Y. (2024). Engineered cellular vesicles displaying glycosylated nanobodies for cancer immunotherapy. Angewandte Chemie International Edition . https://doi.org/10.1002/anie.202404889
Sun, S., Ding, Z., Yang, X., Zhao, X., Zhao, M., Gao, L., Chen, Q., Xie, S., Liu, A., Yin, S., & Xu, Z. (2021). Nanobody: A small antibody with big implications for tumor therapeutic strategy. International Journal of Nanomedicine, 16 , 2337. https://doi.org/10.2147/IJN.S297631
Rolin, C., Zimmer, J., & Seguin-Devaux, C. (2024). Bridging the gap with multispecific immune cell engagers in cancer and infectious diseases. Cellular & Molecular Immunology, 21 (7), 643–661. https://doi.org/10.1038/s41423-024-01176-4
Tapia-Galisteo, A., Compte, M., Álvarez-Vallina, L., & Sanz, L. (2023). When three is not a crowd: Trispecific antibodies for enhanced cancer immunotherapy. Theranostics, 13 (3), 1028–1041. https://doi.org/10.7150/thno.81494
Xu, Z., Gao, C., Jian, M., & Du, W. (2023). Construction of multi-specific antibody by genetic engineering and its progress in tumor therapy. Journal of Biosciences and Medicines, 11 (03), 127–135. https://doi.org/10.4236/jbm.2023.113013
Park, J. A., & Cheung, N. K. V. (2022). Overcoming tumor heterogeneity by ex vivo arming of T cells using multiple bispecific antibodies. Journal for Immunotherapy of Cancer, 10 (1), e003771. https://doi.org/10.1136/jitc-2021-003771
Kang, J., Sun, T., & Zhang, Y. (2022). Immunotherapeutic progress and application of bispecific antibody in cancer. Frontiers in Immunology, 13 , 1020003. https://doi.org/10.3389/fimmu.2022.1020003
Foss, S., Sakya, S. A., Aguinagalde, L., Lustig, M., Shaughnessy, J., Cruz, A. R., Scheepmaker, L., Mathiesen, L., Ruso-Julve, F., Anthi, A. K., & Gjølberg, T. T. (2024). Human IgG Fc-engineering for enhanced plasma half-life, mucosal distribution and killing of cancer cells and bacteria. Nature Communications, 15 (1), 2007. https://doi.org/10.1038/s41467-024-46321-9
Abdeldaim, D. T., & Schindowski, K. (2023). Fc-engineered therapeutic antibodies: Recent advances and future directions. Pharmaceutics, . https://doi.org/10.3390/PHARMACEUTICS15102402/S1
Böldicke, T. (2022). Therapeutic potential of intrabodies for cancer immunotherapy: Current status and future directions. Antibodies (Basel Switzerland), 11 (3), 49. https://doi.org/10.3390/antib11030049
Garattini, L., & Padula, A. (2019). Precision medicine and monoclonal antibodies: Breach of promise? Croatian Medical Journal, 60 (3), 284. https://doi.org/10.3325/CMJ.2019.60.284
Khazaei, M., Hosseini, M. S., Haghighi, A. M., & Misaghi, M. (2023). Nanosensors and their applications in early diagnosis of cancer. Sensing and Bio-Sensing Research, 41 , 100569. https://doi.org/10.1016/J.SBSR.2023.100569
Musnier, A., Dumet, C., Mitra, S., Verdier, A., Keskes, R., Chassine, A., Jullian, Y., Cortes, M., Corde, Y., Omahdi, Z., & Puard, V. (2024). Applying artificial intelligence to accelerate and de-risk antibody discovery. Frontiers in Drug Discovery, 4 , 1339697. https://doi.org/10.3389/FDDSV.2024.1339697
Sun, H., Hu, N., & Wang, J. (2022). Application of microfluidic technology in antibody screening. Biotechnology Journal, 17 (8), 2100623. https://doi.org/10.1002/biot.202100623
Al-wdan, O. A., Sharallah, O. A., Abdelwahab, N. A., Mohammed, A. O., Elmowafy, E., & Soliman, M. E. (2023). Insights into microfabrication and implementation of microfluidics in pharmaceutical drug delivery and analysis. OpenNano, 12 , 100156. https://doi.org/10.1016/J.ONANO.2023.100156
Download references
Acknowledgements
The authors would like to acknowledge Bennett University, India for providing the institutional seed grant to VV and Ph.D. fellowship to AK, and the Department of Science and Technology - Science and Engineering Research Board (DST-SERB) for providing a Start-up Research Grant (SRG/2022/000486) to VV.
This work was supported by institutional seed grant by Bennett University and Start-up Research Grant by the Department of Science and Technology - Science and Engineering Research Board (DST-SERB) (SRG/2022/000486) to Vaishali Verma.
Author information
Authors and affiliations.
Department of Biotechnology, School of Engineering and Applied Sciences, Bennett University, Greater Noida, 201310, Uttar Pradesh, India
Abhavya Raja, Abhishek Kasana & Vaishali Verma
You can also search for this author in PubMed Google Scholar
Contributions
Idea was conceived by VV; the literature search, data analysis and preparation of first draft of the manuscript was done by AR and VV, manuscript draft was edited by AK and VV. All authors read and approved the final manuscript.
Corresponding author
Correspondence to Vaishali Verma .
Ethics declarations
Conflict of interest.
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Raja, A., Kasana, A. & Verma, V. Next-Generation Therapeutic Antibodies for Cancer Treatment: Advancements, Applications, and Challenges. Mol Biotechnol (2024). https://doi.org/10.1007/s12033-024-01270-y
Download citation
Received : 04 June 2024
Accepted : 24 August 2024
Published : 02 September 2024
DOI : https://doi.org/10.1007/s12033-024-01270-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Antibody-drug conjugates
- Bispecific antibodies
- Therapeutics
- Find a journal
- Publish with us
- Track your research
- Cancer Sequencing Methods Overview Cancer Whole-Genome Sequencing Cancer Exome Sequencing Cancer RNA Sequencing Epigenetics
- Cancer Research Applications Overview Liquid Biopsy Research Cancer Single-Cell Analysis Immuno-Oncology Research
Cancer Sequencing Methods
Achieve cancer research insights, combine next-generation sequencing methods for a deeper understanding of cancer.

Utilizing next-generation sequencing in cancer research
NGS-based cancer sequencing methods have expanded our understanding of cancer development, regulation, and progression to unlock new pathways for research. These techniques help detect changes in the cancer genome and identify their impact on the transcriptome, epigenome, and proteome.
Unlike other methods, such as PCR and Sanger sequencing, NGS has the ability to assess thousands of targets at once, greatly amplifying the discovery potential per sample. NGS can also detect low-frequency molecular events associated with carcinogenesis, cancer growth, and metastasis that could be missed using traditional molecular methods. Together, these advancements are paving the future towards improving translational medicine and therapies.
General NGS approaches to cancer research
Researchers can leverage several approaches when studying biological “omes”: bulk-cell analysis, single-cell analysis, spatial analysis, and metagenomic analysis. Each method studies cancer at a different level of biological resolution and has a distinct use case depending on the research goals and objectives.
Bulk-cell analysis : Bulk-cell analysis allows scientists to study pooled cell populations, tissue sections, or biopsies.
Single-cell analysis : Single-cell analysis studies a given “ome” at the resolution of a single cell.
Spatial analysis (also called spatial genomics): Spatial analysis captures “omic” information at the cellular level within an intact tissue sample to link structure and activity.
Metagenomic analysis : Metagenomic analysis sequences every gene in every organism of a complex microbial community present within a tissue, organ, or tumor.
Multiomics in cancer research
Multiomics (multiple omics) integrates data across genomics, transcriptomics, epigenetics, and proteomics to make insights into complex diseases such as cancer. This comprehensive approach aids researchers in understanding molecular changes driving normal development, cellular responses, and diseases.
Learn more about multiomics

Targeted vs. untargeted NGS methods for cancer research
Both targeted and untargeted approaches play crucial roles in research. Targeted research contains some bias since specific, preconceived pathways/targets are assessed. Although this approach may be more efficient and cost-effective in limited cases, untargeted approaches can maximize novel discoveries as they are not dependent on prior knowledge of sequence information. To achieve untargeted sequencing, NGS offers a comprehensive selection of methods to analyze complex cancer samples.
Key cancer sequencing methods
| Method | Description and Use |
|---|---|
| Genomics | |
| Identifies a comprehensive list of cancer-driving genetic events. | |
| A cost-effective and efficient sequencing method to find cancer-driving genes within the coding region of the genome compared to whole-genome sequencing. | |
| Used as a potential alternative to invasive tissue biopsies to detect cell-free circulating tumor DNA (ctDNA), which can act as a noninvasive cancer biomarker. | |
| Targets known DNA and/or RNA variants from the same FFPE sample. | |
| Epigenomics | |
| Both genome-wide analysis and targeted approaches can provide insight into methylation patterns at the single nucleotide level. | |
| Identifies cellular biomarkers associated with regulation of cancer genes or drug resistance. | |
| Determines chromatin accessibility across the genome without prior knowledge of regulatory elements. | |
| Transcriptomics | |
| Measures the average RNA expression and transcriptome changes in cancer samples. | |
| Analyzes gene expression within the natural tumor microenvironment and architecture. | |
| Measures gene expression and explores the distinct biology of individual cancer cells in complex tissues. | |
| Proteomics + Transcriptomics | |
| Uses oligonucleotide-labeled antibodies to simultaneously measure proteins and RNA in single cells. This combined proteomics/transcriptomics approach links RNA expression to cancer phenotypes. | |

Cancer Research Methods Guide
The Cancer Research Methods Guide is a 40+ page comprehensive resource with simple, comprehensive workflows for a broad range of cancer research applications. This guide includes single-cell sequencing, spatial sequencing, methylation profiling, multiomics, cell-free RNA sequencing, and more.

Cancer research applications
Learn about specific cancer research applications and explore resources covering research developments, guides, products, and more.
Sequencing workflow resources
Sequencing platforms.
Our innovative platforms deliver exceptional data quality and accuracy at a massive scale. View sequencer comparison tables and find tools designed to help you choose the right platform for your needs.
Cancer research products
View NGS and microarray products supporting multiple cancer research applications, including tools for analyzing DNA, RNA, epigenetics, and more.
Sequencing data analysis
User-friendly Illumina tools ease the process of analyzing sequencing data so you can spend more time doing research and less time configuring workflows.
Additional cancer research resources
Multiomics methods guide ebook.
This Methods Guide provides examples of multiomic research from recent literature and detailed, end-to-end workflows. Includes recommendations for sample isolation, library prep, sequencing depth, data analysis, and more.
Liquid Biopsy Methods Guide eBook
This 20+ page eBook provides published, comprehensive workflows to thoroughly characterize liquid biopsy samples using NGS and microarrays.
Redefining NGS in cancer research
In this webinar, experts cover essential background topics in NGS, achievements, challenges, and how an integrated multiomics approach can be used in cancer diagnosis and treatment.
Advancing cancer research with multiomics
Learn how researchers at the Ontario Institute for Cancer Research and United Health Network are linking the causes and consequences of complex phenotypes through multiomics to enable discoveries that weren’t possible before.
NGS for beginners
Learn the basics of next-generation sequencing and find tips for getting started.
Interested in getting more information on cancer sequencing methods?
- Current Article
Purdue-invented tool reveals untapped efficacy of cancer drug, leveling up cancer fight

WEST LAFAYETTE, Ind. — Research from Purdue University in collaboration with the National Institutes of Health (NIH) reveals that a cancer drug dismissed by traditional testing methods may be effective in treating bladder cancer, thanks to a new computational tool.
Called the Pathway Ensemble Tool (PET) and described in research published Aug. 24 in Nature Communications , it accurately identifies the most important biological pathways disrupted in cancer and other complex diseases, enabling precise treatment strategies. Biological pathways are the series of steps that molecules take within cells to accomplish specific tasks such as cell growth or energy production.
PET is a novel combination of multiple existing techniques. It was developed by Majid Kazemian, associate professor of biochemistry in the College of Agriculture and computer science in the College of Science at Purdue and member of the Purdue Institute for Cancer Research (PICR). Kazemian worked with Behdad Afzali, the Earl Stadtman Investigator at the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH.
“We conduct large studies using massive amounts of experimental data to determine how well different tools can find pathways related to diseases such as cancer,” Kazemian said. “Surprisingly, we found that the commonly used tools — even though people have tried to make them work well — didn’t do as well as expected in finding important cancer-related pathways. We developed PET, which statistically combines optimized versions of different tools to get much better results. It’s robust and reliable, and it helps us accurately determine which pathways are malfunctioning in cancer without bias, enabling more precise drug identification.”
The research teams used PET to identify dysfunctional biological pathways in 12 different types of cancer. They discovered numerous pathways associated with individual cancers as well as several that signaled either a high or low risk of cancer progression, effectively acting as potential biomarkers of outcomes.
“These biomarkers are crucial for selecting appropriate treatments because they signal when someone has a high risk of death from cancer, even if detected early, and can serve as drug targets,” Kazemian said. “We used PET-derived pathways for drug prediction and discovered effective known and novel drugs that were previously missed using the standard methods of analysis.”
The researchers found that pathways disrupted in bladder and cervical cancers share many genes affected by the enzyme CDK9. When they tested a drug that blocks CDK9, they found that the drug inhibits bladder and cervical cancer cell growth in lab and animal tests more effectively than previous evaluation tools had indicated.
In a test specific to bladder cancer, PET determined that the CDK9-inhibitor drug called CCT068127 was more effective at slowing cancer-cell growth than other drugs tested. This finding was unexpected, as previous researchers had not identified CDK9 as a target for bladder cancer. The finding has led to a new clinical trial involving fellow PICR affiliate Deborah Knapp, Distinguished Professor of Comparative Oncologyat Purdue. She is working to study the efficacy of the clinically available CDK9 inhibitor in dogs with bladder cancers that are similar to human bladder cancers.
“We anticipate that our findings will allow for the development of improved pathway discovery tools similar to PET,” Kazemian said. “This will lead to tangible insights into disease mechanisms and identify novel diagnostic markers and prognostic markers, and therapeutics for cancer.”
The study, titled “Unbiased discovery of cancer pathways and therapeutics using Pathway Ensemble Tool and Benchmark,” was published in Nature Communications, a multidisciplinary open-access journal dedicated to publishing high-quality research in the biological, physical, chemical and Earth sciences.
About Purdue University
Purdue University is a public research institution demonstrating excellence at scale. Ranked among top 10 public universities and with two colleges in the top four in the United States, Purdue discovers and disseminates knowledge with a quality and at a scale second to none. More than 105,000 students study at Purdue across modalities and locations, including nearly 50,000 in person on the West Lafayette campus. Committed to affordability and accessibility, Purdue’s main campus has frozen tuition 13 years in a row. See how Purdue never stops in the persistent pursuit of the next giant leap — including its first comprehensive urban campus in Indianapolis, the Mitch Daniels School of Business, Purdue Computes and the One Health initiative — at https://www.purdue.edu/president/strategic-initiatives .
Media contact: Amy Raley, [email protected]
Source: Majid Kazemian, [email protected]
- Open access
- Published: 02 September 2024
Multibiomarker panels in liquid biopsy for early detection of pancreatic cancer – a comprehensive review
- Kim-Lea Reese 1 ,
- Klaus Pantel 1 &
- Daniel J. Smit 1
Journal of Experimental & Clinical Cancer Research volume 43 , Article number: 250 ( 2024 ) Cite this article
3 Altmetric
Metrics details
Pancreatic ductal adenocarcinoma (PDAC) is frequently detected in late stages, which leads to limited therapeutic options and a dismal overall survival rate. To date, no robust method for the detection of early-stage PDAC that can be used for targeted screening approaches is available. Liquid biopsy allows the minimally invasive collection of body fluids (typically peripheral blood) and the subsequent analysis of circulating tumor cells or tumor-associated molecules such as nucleic acids, proteins, or metabolites that may be useful for the early diagnosis of PDAC. Single biomarkers may lack sensitivity and/or specificity to reliably detect PDAC, while combinations of these circulating biomarkers in multimarker panels may improve the sensitivity and specificity of blood test-based diagnosis. In this narrative review, we present an overview of different liquid biopsy biomarkers for the early diagnosis of PDAC and discuss the validity of multimarker panels.
The most common type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC), which accounts for more than 90% of all pancreatic cancers [ 1 ]. PDAC-related precancerous conditions include pancreatic intraepithelial neoplasms (PanINs), intraductal papillary mucinous neoplasms (IPMNs), and mucinous cystic neoplasms (MCNs). The etiology of pancreatic cancer is not fully understood, but several risk factors are associated with PDAC. In addition to common cancer risk factors, including age, obesity, genetic predispositions, smoking and alcohol consumption, the factors conferring the highest risk are type 2 diabetes mellitus and chronic pancreatitis [ 2 ].
Pancreatic cancer is the seventh most commonly diagnosed cancer and the fourth most frequent cause of cancer-related deaths in Europe; it accounted for almost as many diagnoses (140,116 cases) as deaths (132,134 deaths) in 2020 [ 3 ]. Despite tremendous efforts in research and new therapies resulting in increased survival rates of patients with other cancer types, pancreatic cancer still has a low 5-year survival rate of approximately 10%, with a median overall survival (OS) of less than six months [ 4 ]. One of the main reasons is late diagnosis, as patients do not show specific early clinical symptoms [ 5 ]. At the time of PDAC detection, less than 20% of tumors are eligible for curative resection [ 6 ]. However, surgery followed by adjuvant systemic chemotherapy is the best therapeutic option, significantly increasing the 5-year survival rate [ 7 ]. At advanced tumor stages with metastases, the only remaining treatment is systemic chemotherapy, which has a low response rate and a high resistance rate [ 6 ]. Consequently, it is highly important to develop diagnostic tests that enable the detection of early-stage PDAC (AJCC/UICC stages I and II) to improve the OS and progression-free survival (PFS) of patients.
PDAC is mainly diagnosed through medical imaging methods, including computer tomography, magnetic resonance imaging, magnetic or endoscopic retrograde cholangiopancreatography, and endoscopic ultrasound-guided fine needle aspiration. However, a clear diagnosis is not always possible because of the retroperitoneal location of the pancreas and the small size of early-stage PDAC lesions [ 6 , 8 ]. In addition, screening for the early diagnosis of PDAC by imaging is not practical, as it is neither cost nor time efficient and involves exposure to radiation [ 8 ].
Liquid biopsy (LB) is a minimally invasive procedure that allows the sampling and analysis of body fluids, thus enabling cancer diagnosis, treatment monitoring, surveillance, and prognostication [ 9 ]. The samples can be obtained from various body fluids, including urine, saliva, cerebrospinal fluid, bone marrow, or blood [ 10 ]. In recent years, blood has been one of the most popular analytes, as blood sampling is easy, cost-effective, and repeatable [ 11 ]. Biomarkers that can be detected in the blood include circulating tumor cells (CTCs), circulating host cells, including cancer-associated fibroblasts (CAFs) or circulating endothelial cells (CECs), circulating cell-free RNA and DNA (cfRNA, cfDNA), extracellular vesicles (EVs) and proteins [ 12 ] ( Fig. 1 ) . However, to date, no single biomarker or multimarker panel can reliably diagnose PDAC, especially in the early stages.

Overview of (blood-based) liquid biopsy analytes for the early detection of pancreatic cancer. The figure was created with BioRender.com under academic license
In this review, we summarize potential biomarkers and detection methods for blood-based liquid biopsy and discuss their implications for the early-stage detection of PDAC. Moreover, we review publications on multibiomarker panels for PDAC diagnosis and highlight their importance in the early diagnosis of pancreatic cancer.
The literature search for this narrative review regarding multibiomarker panels was carried out in PubMed on 4th July 2024 via the following terms:
("biomarker panel*" OR "marker panel*" OR "multi biomarker*" OR "multi marker*" OR "marker combin*" OR "biomarker combin*") AND (pancrea*) AND (cancer OR carcinoma OR tumor* OR adenocarcinoma*) AND (sera OR serol* OR plasma OR blood OR "liquid biops*" OR "fluid biops*").
The literature search was restricted to articles in English and yielded 126 papers. Reviews and case reports were excluded, as were studies that did not involve research on humans, PDAC, diagnosis, or blood-based liquid biopsy. According to these criteria, a total of 57 publications were further analyzed for this review. A flow chart can be found in Fig. 2 . The data extracted from the publications included study details (author, year of publication, country), biomarker details (biomarkers used, detection method, fluid type), patient cohort details (patient numbers, stage, controls), and statistical details (sensitivity, specificity, area under the curve (AUC)). If separate data for early-stage PDAC (pathological AJCC/UICC stages I and II according to the 8th edition of the staging manual) and late-stage or all-stage PDAC diagnosis were provided, only the values for early-stage PDAC were included.

Overview of the results from the literature search and selection of included studies
Liquid biopsy biomarkers for PDAC
Currently used tumor markers for pdac.
A commonly used serological biomarker for PDAC is carbohydrate antigen 19–9 (CA19-9), also referred to as sialyl Lewis-A, which currently represents the only FDA-approved marker. Increased levels of CA19-9 have been reported in PDAC patients compared with healthy individuals [ 13 , 14 ]. The concentration of CA19-9 and its sensitivity as a diagnostic marker increase with increasing PDAC stage [ 15 , 16 ], with the most pronounced increase detected between AJCC/UICC stage II and stage III. However, especially in early stages (e.g., stage I [ 17 ]), the level of CA19-9 is similar to that in various benign conditions, precancerous lesions, and other malignancies (e.g., colorectal cancer, gastric cancer, hepatocellular carcinoma), resulting in low specificity [ 14 , 15 ]. With respect to its use for diagnostic purposes, it is important to consider that 6% of Caucasians and 22% of non-Caucasians who lack Lewis antigen A cannot produce CA19-9, subsequently leading to false-negative results [ 18 ]. Consequently, international guidelines do not recommend its use as a diagnostic method but rather as a longitudinal marker in patients with detectable CA19-9 at baseline [ 16 ]. Owing to the lack of robust biomarkers, various markers have been investigated as possible candidates with increased sensitivity and specificity for PDAC diagnosis. The following sections present an overview of cellular and acellular liquid biopsy-based biomarkers for PDAC diagnosis.
Cellular biomarkers
One group of biomarkers analyzed in liquid biopsy is cells that have detached from their site of origin and entered the bloodstream. These can be derived from tumor or noncancerous host cells that are part of the tumor microenvironment (TME) (e.g., immune cells, fibroblasts, and endothelial cells) [ 19 ].
Tumor cells detected in the blood are referred to as circulating tumor cells (CTCs) [ 9 ]. CTCs are highly heterogeneous even if derived from the same patient [ 20 ]. Compared with classical diagnostic biopsies (e.g., fine needle biopsy), CTCs are able to provide a more representative image of tumor heterogeneity [ 21 , 22 , 23 ]. However, detecting CTCs is still challenging, as approximately one CTC is detectable among more than a million other blood cells (e.g., erythrocytes, leukocytes, and platelets), and CTCs have a short half-life of only 1–2.4 h [ 24 , 25 ]. In addition to the number of CTCs, their genome, transcriptome, proteome and functional properties can be analyzed [ 26 ].
With respect to PDAC, a meta-analysis of 19 studies revealed that more than half of patients (707 out of 1320 patients analyzed) had detectable CTCs in their blood [ 27 ]. These patients had lower OS and PFS rates than CTC-negative patients did, highlighting the adverse prognostic effect of CTCs in PDAC patients. However, most patients in the studies included in the meta-analysis were in advanced tumor stages (stage III and IV: 61%), with only 31% in stage II and only 8% in stage I. The low number of CTCs, particularly in early PDAC stages, may lead to false negative results and low sensitivity [ 12 , 28 ]. A potential explanation for the low CTC number in PDAC could be the filtration of CTCs in the liver before they reach the peripheral blood vessels and the reduced blood flow within the cancerous pancreas [ 29 , 30 ]. This limits the analysis of CTCs as possible biomarkers for early diagnosis, but with the emergence of novel, more sensitive analysis techniques (e.g., in vivo CTC capture devices [ 31 ]) and techniques that allow processing of larger volumes [ 32 , 33 ], this limitation may be overcome [ 34 ]. Despite the impaired sensitivity that could arise from the different methods used or the heterogeneity of CTCs, the specificity of CTCs for PDAC diagnosis has been reported in several studies to reach > 90% [ 35 , 36 , 37 ].
In PDAC, cells make up only a small part of the tumor, while the largest part is the dense stroma that forms the tumor microenvironment [ 38 ]. Compared with other solid tumors, PDAC has the most pronounced desmoplastic stroma reaction, which generates a physical barrier around the tumor, thereby impairing radical resection and increasing therapy resistance [ 39 , 40 ]. Although the composition and structure of the stroma varies between patients, it consists of several main components [ 41 ]. Noncellular components, including glycoproteins, fibronectins, collagens, and enzymes, form the extracellular matrix (ECM). The cellular components include endothelial, immune, and stromal cells, including pericytes, and local cancer-associated fibroblasts (CAFs). These host cells can also detach from the TME, enter the bloodstream, and be analyzed as possible liquid biopsy biomarkers for PDAC (e.g., as circulating CAFs (cCAFs)).
CAFs are key components of the TME, and they are near or in direct contact with cancer cells [ 38 ]. The three different major types of CAFs, myofibroblast CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs), are associated with distinct functions and phenotypes [ 42 ]. These functions include the production of cytokines, chemokines, metabolites, enzymes, and ECM molecules to prevent or promote tumor growth [ 43 ]. cCAFs are found in the blood of patients with various tumors, including PDAC, where they are linked to a poorer prognosis in advanced stages [ 44 , 45 , 46 ]. Only one study examined cCAFs in six PDAC patients and reported an association between the presence of cCAFs and poorer clinical outcomes as well as lower OS rates at metastatic stages [ 47 ]. However, to our knowledge, there are currently no studies on the role of cCAFs in the early stages of PDAC, and such studies are crucial for identifying their suitability as biomarkers for early PDAC diagnosis.
Circulating nucleic acids
Circulating tumor DNA (ctDNA) is a type of cell-free DNA (cfDNA) derived from tumor cells that can be found in the bloodstream. ctDNA can be released by cells undergoing apoptosis and necrosis or can be actively transported through the cell membrane [ 48 ]. Since only up to 1% of the cfDNA in the blood of early-stage patients originates from tumors, most detectable circulating nucleic acids are cfDNA from noncancer cells, which limits the ability to detect ctDNA [ 49 , 50 ]. The amount of ctDNA in the blood varies between different tumors and increases up to 40% in advanced tumor stages [ 50 , 51 ]. The half-life of ctDNA is estimated to be between 16 and 114 min, which makes isolation more challenging [ 52 , 53 ]. ctDNA can be detected due to specific alterations in the tumor and can be examined for mutations, DNA integrity, gene fusion, copy number variation, or methylation status [ 54 , 55 ]. The high concordance between mutations in ctDNA and those in tumor tissue makes it suitable as a biomarker that provides information about the primary tumor even if it is inaccessible [ 56 ].
Analysis of genomic aberrations in all-stages PDAC tissue revealed a panel of four genes, namely, KRAS , CDKN2A , TP53, and SMAD4, with mutation frequencies of 90%, 90%, 70%, and 55%, respectively, as the main genomic drivers of PDAC [ 57 , 58 , 59 , 60 , 61 ]. Interestingly, mutations in these genes can be detected in preneoplastic PanIN lesions; notably, KRAS is the first event, and subsequent alterations in CDKN2A , TP53, and SMAD4 can be detected in higher-grade PanINs [ 62 , 63 ]. These mutations lead to increased proliferation, dysregulation of the cell cycle and an impaired DNA damage response [ 59 ]. As KRAS mutations are among the initiating mutations during the development of PDAC, KRAS mutations are interesting biomarkers for the early diagnosis of PDAC [ 64 ]. On the basis of the molecular profile of PDAC, several studies have used these mutations for ctDNA detection [ 64 ]. However, germline mutations in cfDNA or clonal hematopoiesis of indeterminate potential (CHIP) in noncancerous cells, especially related to KRAS (approximately 30%) [ 65 ], may lead to false-positive results and should be considered in related evaluations [ 55 , 66 , 67 ].
A meta-analysis of seven retrospective studies on the utility of ctDNA as a liquid biopsy biomarker revealed a sensitivity of 64% (95% CI 0.58–0.70), a specificity of 92% (95% CI 0.88–0.95), and an AUC of 0.9478 across all PDAC stages [ 28 ]. With approximately only one molecule of ctDNA in every 5 mL of plasma, the moderate sensitivity is presumably the result of minute amounts of released ctDNA, especially in the early tumor stages, when the rates of apoptosis and necrosis are lower [ 68 , 69 ].
In addition to somatic cancer alterations, epigenetic traits (e.g., methylation, fragmentation) can also be examined in ctDNA. Epigenetic alterations have recently received much attention, as they may also provide tissue-specific information that helps to determine the organ in which the tumor originates. Nicholson et al . analyzed the cfDNA methylation pattern in a prospective study of 5,461 participants with suspected cancer and were able to detect different tumors with a sensitivity of 66.3% in all stages and 24.4% in stage I patients, with a specificity of 98.4% [ 70 ]. A recent publication by García-Ortiz et al . reviewed studies analyzing the ctDNA methylation status in PDAC and concluded that the use of a single epigenetic biomarker does not allow for the diagnosis of early-stage PDAC and suggested that a multimarker panel would be more efficient [ 71 ]. Moreover, fragmentomic approaches focusing on fragment size, fragment ends, and end motifs can reveal differences between ctDNA and cfDNA [ 72 ]. ctDNA from cancer patients is shorter than nontumor cfDNA, and its feautures differ between different tumor entities, which enables the identification of the tissue of origin [ 73 , 74 ]. Cristiano et al. were able to detect pancreatic tumors with a sensitivity of 71% at a specificity of 95% on the basis of the cfDNA fragment size [ 54 ]. These studies underscore the promising value of cfDNA-based approaches that are independent of the presence of genomic signatures.
In addition to cfDNA, cancer cells also release cell-free RNA (cfRNA) into the circulation [ 75 , 76 ]. In addition to intracellular coding messenger RNAs (mRNAs), which are required for protein synthesis, noncoding RNAs, including microRNAs (miRNAs), are potential biomarker candidates [ 77 ]. cfRNAs are highly stable, as they are typically packed in extracellular vesicles or attached to lipid or protein complexes rather than circulating freely in the bloodstream [ 78 , 79 , 80 , 81 , 82 ].
miRNAs are noncoding, single-strand RNAs with an average length of 22 nucleotides that are highly evolutionarily conserved among various species [ 83 ]. miRNAs can regulate their target mRNAs at the posttranscriptional level by affecting their translation and stability [ 84 , 85 , 86 ]. In cancer patients, altered expression of miRNAs has been reported [ 87 ]. Numerous studies examining the role of various miRNAs as LB biomarkers in PDAC have been conducted and reviewed elsewhere [ 57 ]. In a meta-analysis, Peng et al. examined miRNAs from 46 studies involving 4,326 patients with pancreatic cancer [ 88 ]. The diagnostic performance of miRNA panels, which included 4.5 miRNAs on average (range: 2–12 miRNAs), was compared to that of single miRNAs and interestingly exhibited no significant diagnostic benefit. The combined results yielded a sensitivity of 79% (0.77–0.81), a specificity of 77% (0.75–0.79), and an AUC of 0.85 (0.81–0.87). Considering only early-stage PDAC (up to stage IIA), the diagnostic value decreased slightly to a sensitivity of 79% (0.76–0.82), a specificity of 74% (0.68–0.79), and an AUC of 0.81 (0.77–0.84) [ 88 ].
Proteins are important for communication between cancer cells and host cells in the TME [ 89 ]. Proteins can be located on the membrane surface of cells but can also be secreted in vesicles or released into the circulation [ 90 ]. A wide range of different circulating proteins, including cytokines, chemokines, carbohydrate antigens, growth factors, inflammatory factors, glycoproteins, and apolipoproteins, orchestrate numerous biological processes [ 91 , 92 , 93 ]. Proteins released by cancer cells can regulate the development and progression of cancer by promoting invasion and metastasis [ 89 ]. Several proteins are up- or downregulated in the blood of PDAC patients compared to that of healthy donors or benign tumor patients [ 94 ]. Hence, many circulating proteins have been analyzed as potential biomarkers for PDAC and are reviewed in more detail elsewhere [ 95 ]. However, interestingly, the sensitivity and specificity of most single proteins do not exceed those of CA19-9 [ 5 ]. A meta-analysis by Kane et al . compared 250 prospective and retrospective studies published before July 2020 on all stages of PDAC; the results revealed an AUC of 0.85 for CA19-9 alone and 0.783 for novel single biomarkers [ 96 ].
Extracellular vesicles
Extracellular vesicles (EVs) are lipid-bound and secreted particles that comprise three classes of vesicles: exosomes (30–150 nm), microvesicles (50–1000 nm), and apoptotic bodies (500–5000 nm). EVs can be released by several cell types, including neurons, epithelial cells, and fibroblasts, as well as cancer cells [ 97 , 98 , 99 ]. Exosomes are particularly interesting for liquid biopsy approaches because they contain many molecules, including lipids, metabolites, nucleic acids (e.g., miRNAs and mRNAs), and proteins, that are protected from degradation by the EV membrane [ 100 ]. As exosomes transfer these molecular cargoes to recipient cells through cell‒cell interactions or even over large distances, e.g., between different organs, they are important for cellular communication [ 101 ].
Exosomes influence tumor malignancy by regulating the tumor microenvironment, angiogenesis, tumor growth, invasion, and metastasis, including epithelial‒mesenchymal transition, immunomodulation, and chemoresistance [ 98 , 102 , 103 , 104 , 105 , 106 ]. Moreover, cancer cells, including PDAC cells, secrete more exosomes than noncancerous cells [ 107 , 108 ].
In a meta-analysis on the potential utility of extracellular vesicle cargo as biomarkers for PDAC, Jia et al. examined 39 studies including 2,037 PC patients [ 109 ]. Seventeen studies on EV RNAs, 16 on EV proteins, and 16 on EV biomarker panels were evaluated across all tumor stages. The most reported molecules were the EV RNAs miR-21 and miR-10b and the EV proteins GPC1 and EphQ2. A sensitivity of 84% (95% CI: 81–86%) and a specificity of 89% (95% CI: 86–91%) were obtained from the pooled values of EV RNAs and EV proteins. In contrast to analysis of the previously described biomarker types, the analysis of EVs in early PDAC stages I and II led to an increased sensitivity of 90% (95% CI: 87–93%) and specificity of 94% (95% CI: 92–95%). Interestingly, EVs as markers seem to perform at least as well and possibly even better in earlier stages (although only modestly with almost overlapping CIs) than in advanced stages [ 109 ]. One potential explanation could be that patients with advanced PDAC suffer from dysregulated EV secretion due to cancer-related effects, including cachexia and dysregulated metabolic processes. These findings indicate that exosomes and their cargo are potential biomarkers for the early diagnosis of PDAC.
Multimarker analysis
Numerous single biomarkers for the diagnosis of PDAC have been investigated, as a single marker can facilitate diagnostic assay development and implementation in routine clinical practice. However, the investigated markers have low sensitivity and specificity for diagnosis. Considering the high degree of patient diversity and tumor heterogeneity, a multimarker panel can provide complementary value and seems to perform better than single biomarkers do.
The literature search yielded 57 papers that analyzed multibiomarker panels in blood to diagnose PDAC. As some publications included two different panels, the total number of multibiomarker panels assessed was 63. Among these panels, 57 included proteins, 10 included RNA, 6 included EVs, 4 included cfDNA, 2 included metabolites, and 1 included CTCs. An overview of these studies [ 16 , 110 , 111 , 112 , 113 , 114 , 115 , 116 , 117 , 118 , 119 , 120 , 121 , 122 , 123 , 124 , 125 , 126 , 127 , 128 , 129 , 130 , 131 , 132 , 133 , 134 , 135 , 136 , 137 , 138 , 139 , 140 , 141 , 142 , 143 , 144 , 145 , 146 , 147 , 148 , 149 , 150 , 151 , 152 , 153 , 154 , 155 , 156 , 157 , 158 , 159 , 160 , 161 , 162 , 163 , 164 , 165 ] can be found in Table 1 .
Many publications started by analyzing single biomarkers and later combined them with one or more other biomarkers. The addition of biomarkers led to increased sensitivity and specificity and improved AUC values in these studies. For example, Capello et al . calculated an AUC of 0.730 for TIMP1, 0.832 for LRG2, and 0.821 for CA19-9 to distinguish early-stage PDAC patients from healthy controls [ 133 ]. The combination of all three protein markers increased the AUC to 0.887, and adding 5 metabolites to the protein panel further increased the AUC to 0.924 [ 161 ]. This observation was quantified in the above-mentioned meta-analysis by Kane et al . [ 96 ]. The pooled AUC for studies with single biomarkers was 0.803, which was significantly lower than the multibiomarker panel AUC of 0.898. However, this analysis was performed on all stages of PDAC and did not focus particularly on the early stages.
The investigated biomarkers were combined with CA19-9 analysis in 55 of the 63 studies, and only 8 studies did not include CA19-9 in their panel [ 129 , 150 , 151 , 152 , 153 , 154 , 155 , 165 ]. Adding CA19-9 to other biomarkers improved the diagnostic power. For example, Dong et al . examined the proteins POSTN and CA242 in early-stage PDAC patients versus healthy controls and reported an AUC of 0.92 for their combination [ 139 ]. The addition of CA19-9 to the panel increased the AUC to 0.98.
Most biomarker panels included only protein markers (46 out of 63 studies) that were analyzed directly from the blood or isolated from EVs. In the studies on early-stage PDAC, the AUCs ranged from 0.76–0.98 (Fig. 3 ). The protein panels consisted of two proteins in 7 studies, three proteins in 11 studies, and four or more proteins in 5 studies, although the number of proteins did not appear to directly correlate with the reported AUC. The biomarker panels included numerous different proteins, whereas only some proteins, including CA19-9, CEA, and MUC5AC, were found in several panels. Hinestrosa et al . isolated EVs from the blood of early PDAC patients and healthy controls and analyzed a panel of 13 proteins within EVs, resulting in a sensitivity of 95.7% and specificity of 99.5% [ 147 ].

Distribution of the AUC values of multibiomarker panels in identifying stage I and II PDAC patients. The AUC values derived from Table 1 for the multibiomarker panels for early-stage PDAC are plotted. The figure includes multimarker panels consisting of combinations of proteins (20 studies), DNA (1 study), RNA (2 studies) or multiomic markers (5 studies)
Only five studies focused on other types of biomarkers, namely, cfDNA [ 150 , 151 ] or RNA [ 152 , 153 , 154 ]. Eissa et al . analyzed the cfDNA methylation pattern of the BNC1 and ADAMTS1 genes and reported the ability to distinguish early-stage PDAC patients from mixed controls, with an AUC of 0.95 [ 150 , 151 ]. Ganepola et al . compared the miRNAs miR-642b, miR-885-5p, and miR-22 between stage II PDAC patients and healthy controls as well as high-risk patients, resulting in an AUC of 0.97 [ 152 ]. A prospective study analyzing the 2’-O-methylated miRNAs miR-28-3p, miR-143-3p, and miR-151a-3p in 135 individuals was performed by Yang et al . The panel identified 20 out of 28 early-stage PDAC patients, resulting in an AUC of 0.81 [ 154 ].
Furthermore, several studies have investigated multiomic panels by combining analyses of CTCs, cfDNA, metabolites, or miRNAs with proteins [ 155 , 156 , 157 , 158 , 159 , 160 , 161 , 162 , 163 , 164 , 165 ]. The only reviewed study that included CTCs for PDAC diagnosis was performed by Chen et al . [ 159 ]. The authors isolated and quantified CTCs from whole blood and added CA19-9 analysis to distinguish all-stage PDAC patients from mixed controls with an AUC of 0.95. Two studies analyzed cfDNA in combination with different proteins: Cohen et al . focused on four proteins (CA19-9, CEA, HGF, OPN) and KRAS mutations in cfDNA, distinguishing early-stage PDAC patients from healthy controls with a sensitivity of 64% and a specificity of 99.5% [ 160 ]. In a different cfDNA analysis approach, Berger et al . quantified ctDNA and analyzed CA19-9 and THBS2 to differentiate between all-stage PDAC, IPMN and pancreatitis, with an AUC of 0.94 [ 157 ]. Metabolites were included in the studies of Fahrmann et al . and Zhao et al . to distinguish early-stage PDAC patients from healthy controls. The multibiomarker panel of Fahrmann et al . consisted of five metabolites (acetylspermidine, diacetylspermine, an indole-derivative, and two lysophosphatidylcholines) and three proteins (CA19-9, LRG1, and TIMP1), resulting in an AUC of 0.924 [ 161 ]. Zhao et al . combined three metabolites (proline, creatine, and palmitic acid) with CA19-9, and this panel had an AUC of 0.949 [ 163 ]. Seven studies involved combined analysis of proteins with cell-free RNAs or miRNAs isolated from EVs [ 155 , 156 , 158 , 162 , 164 , 165 ]. Nakamura et al . combined CA19-9 with 5 cell-free miRNAs (miR30c-5p, miR340-5p, miR335-5p, miR23b-3p, and miR142-3p) and 8 EV-derived miRNAs (miR145-5p, miR200b-3p, miR429, miR1260b, miR145-3p, miR216b-5p, miR200a-3p, and miR217-5p) [ 162 ]. This biomarker panel had an AUC of 0.99 for distinguishing early-stage PDAC patients from healthy controls, indicating that it was the most precise diagnostic panel among all reviewed studies on early-stage PDAC. However, there were some limitations to this study, such as the modest sample size ( n = 91) and the lack of age-matched control groups, which need to be addressed before the biomarker panel can be applied in the clinic.
The analyzed biomarkers were tested on early-stage PDAC patient samples in 34 of the 63 studies and had AUCs in the range of 0.76–0.99 (Fig. 3 ). The protein panels had the lowest mean AUC of 0.89, and the range of AUC values was the widest. The multiomic panels had the highest mean AUC of 0.95, with a small range from 0.92–0.99, indicating that combining different omic biomarkers yields greater statistical power. However, compared with single markers or panels with only one type of marker (e.g., protein), multimarker panels, particularly multiomic panels, involve more elaborate integrative assays with potentially increased development time and greater complexity.
These studies indicate that several biomarkers perform well in detecting early-stage PDAC. However, for these markers to be used for the screening of risk groups, the sensitivity and specificity need to be increased to minimize the number of false positive and negative diagnoses. In particular, high sensitivity is difficult to reach, as PDAC shares numerous biomarkers and mutations (e.g., RAS mutations) with other diseases (e.g., colorectal cancer), benign diseases of the pancreas (e.g., pancreatitis) or its precancerous lesions (e.g., IPMN), and these other diseases have higher prevalence in the general population than PDAC [ 166 , 167 ]. Diseased controls (e.g., those with precancerous conditions, pancreatitis, and pancreatic cysts) and individuals at risk for developing PDAC should be included in studies to minimize false-positive rates and gain further knowledge of the molecular tumorigenesis of PDAC. On the basis of these assumptions, screening for PDAC in the general population could be implemented in pancancer screening efforts rather than as a specific test for PDAC. A panel of multiple markers could be used to screen for several cancer types at the same time and therefore be used on a broader group of individuals. Two multimarker panel-base tests, CancerSEEK [ 168 ] and Galleri (GRAIL) [ 169 , 170 , 171 , 172 ], were developed to detect the early stages of multiple tumors, including PDAC. The CancerSEEK multimarker panel includes ctDNA and eight proteins (CA-125, CEA, CA19-9, PRL, HGF, OPN, MPO, and TIMP-1) to detect ovarian, liver, stomach, pancreatic, esophageal, colorectal, lung and breast tumors. Currently, the CancerSEEK test is only used in clinical trials (NCT04213326). The Galleri test by GRAIL, which is already commercially available, analyzes the whole-genome methylation of cfDNA to detect signals of more than 50 cancer types. In an independent validation set of 4,077 individuals, the test was able to identify 35 of 41 patients with early-stage pancreatic cancer, resulting in a sensitivity of approximately 60% at 99.5% specificity [ 171 ]. Moreover, in another prospective study of 6,662 participants, the Galleri test detected a suspicious positive cancer signal in 92 cases [ 172 ]. After 12 months of follow-up, 35 of these 92 participants (38%) were confirmed to have a true positive cancer diagnosis, and 6,235 of the 6,549 (95.5%) participants without a cancer signal had true negative results, highlighting the feasibility of multicancer early detection (MCED) testing. Moreover, for the first time, this study assessed the subsequent diagnostic pathways and time to diagnostic resolution [ 172 ]. Notably, these studies by Schrag et al., are highly important for emphasizing the clinical utility of screening approaches (e.g., MCED tests), even in nonrisk groups.
Another study on the methylation of cfDNA, the THUNDER study, yielded high sensitivity for advanced stages but only approximately 35% sensitivity for stages I and II, with 98.9% specificity for detecting pancreatic tumors [ 173 ]. Although the specificity of the tests is sufficient, the sensitivity for early-stage diagnosis is lower than that of the PDAC-specific panels outlined in this review and therefore indicates that high-risk groups may benefit from a PDAC-specific screening approach. Moreover, the number of false-negative results leads to high costs for further diagnosis and insecurities for tested individuals. However, for some cancer types, the sensitivity reached relatively high values (e.g., 100% (CancerSEEK), 100% (Galleri), and 75% (THUNDER) for diagnosing stage I liver cancer). With respect to the diagnosis of advanced stages, the sensitivity was 80% to 100% for several cancer types in all tests. Further investigations on early-stage cancers are necessary to establish a reliable multicancer test, which would greatly improve the diagnosis of cancer.
In many of the reported PDAC studies in this review, several limitations affected the results of the analyses. Only 34 of the 63 studies assessed the analyzed biomarkers in early-stage PDAC patient samples. Notably, surgery is most efficient treatment in the early stages of PDAC and can significantly improve the OS of patients [ 5 , 174 , 175 ]. Thus, studies focusing on samples of early-stage patients are urgently needed to find suitable biomarkers for early diagnosis. Moreover, many studies analyzing single or multiple biomarkers had small samples sizes, resulting in low statistical power. Considering the high heterogeneity of patients and tumors, large cohorts (optimally from multiple centers) are needed to reliably assess biomarkers. Another limitation of most studies is the retrospective study design. Although many samples were collected prospectively, the analysis was performed retrospectively on chosen samples. Only three biomarker panels were tested in a prospective study [ 115 , 129 , 176 ], yielding a higher level of medical evidence. Another important aspect for future studies is the standardization of preanalytical factors as well as methods used for detection. Multicenter evaluation of CTCs, DNAs, and miRNAs in standardized blood samples revealed significant differences between the technologies used at different centers [ 177 , 178 , 179 ], which prevents the comparison of results and thereby limits the development of novel diagnostic assays. Therefore, it is necessary to use standardized protocols for the handling of samples and the performance of assays to improve the quality of studies. Several international liquid biopsy consortia and societies, including the European Liquid Biopsy Society (ELBS) and the International Liquid Biopsy Standardization Alliance (ILSA), are currently collaborating on this task. The establishment of reference and uniform cutoff values for promising biomarkers is important for performing randomized prospective studies to obtain more robust medical evidence. These prospective studies must be hypothesis-driven and have defined enrollment criteria to avoid the risk of missing many specific features related to the complex clinical condition of pancreatic cancer to address unmet needs. Moreover, after these studies have been completed, it is highly important to systematically review or conduct a meta-analysis of the available studies to select the best features and guide further diagnostic assay development.
Conclusion and perspectives
This review of studies on using multimarker panels in blood samples to diagnose PDAC revealed that the use of panels of multiple biomarkers compared with single biomarkers improved the diagnostic power. Most studies have been performed on protein panels, whereas only a few have analyzed other biomarker types or even multiomic panels. Although many biomarkers had low diagnostic power alone, the combination of these biomarkers with CA19-9 increased the diagnostic power; thus, CA19-9 is part of many multibiomarker panels. For future studies, it is crucial to conduct prospective studies with standardized methods and to use samples of patients in the early stages to enable the development of a biomarker panel for early-stage PDAC diagnosis, allowing early therapeutic intervention. Owing to the low number of early-stage PDAC patients, collaborative efforts in a multicenter setting are needed. Currently, 361 ongoing studies on early-stage diagnosis of pancreatic cancer are listed at clinicaltrials.gov, illustrating the unmet need for reliable early detection methods. More than 40% of these ongoing studies (152 studies) include liquid biopsy-based diagnostics, and four of these liquid biopsy-based studies are MCED tests that utilize DNA methylation or multiomic panels. One of these recent liquid biopsy-based studies was initiated by the EU-funded PANCAID consortium [ 180 ]. Researchers in the PANCAID consortium have committed their research toward finding novel minimally invasive multimarker panels for early PDAC detection. Findings related to the early-stage detection of primary disease may also apply to the early diagnosis of minimal residual disease in cancer patients who have undergone treatment with curative intent after diagnosis [ 181 , 182 ]. The European Consortium GUIDE.MRD is currently tackling this ambitious task for the detection of ctDNA in patients with pancreatic, colorectal, and lung cancers [ 183 ].
Availability of data and materials
Not applicable.
Abbreviations
Acute pancreatitis
Antigen-presenting CAFs
Area under the curve
Benign disease
Carbohydrate antigen 19–9
Cancer-associated fibroblast
Circulating CAF
Cell-free DNA
Cell-free RNA
Clonal hematopoiesis of indeterminate potential
Chronic pancreatitis
Circulating tumor cell
Circulating tumor DNA
Diabetes mellitus
Electrochemiluminescent-based immunoassays
Enzyme immunoassay
Enzyme-linked immunosorbent assay
Extracellular vesicle
Healthy control
Immunoassay
Individuals at risk
Inflammatory CAF
Intraductal papillary mucinous neoplasm
- Liquid biopsy
Multi-cancer early detection
Mucinous cystic neoplasm
Magnetic immunoassay
Methylation on beads
Messenger RNA
Mass spectrometry
Myofibroblast CAF
Overall survival
Pancreatic intraepithelial neoplasm
Pancreatic cyst
Polymerase chain reaction
Pancreatic ductal adenocarcinoma
Progression-free survival
Proximity ligation assay
Radioimmunoassay
Sensitivity
Specificity
Tumor microenvironment
Liou GY, Byrd CJ. Diagnostic bioliquid markers for pancreatic cancer: what we have vs. what we need. Cancers (Basel). 2023;15(9):2446.
Article PubMed CAS Google Scholar
Maisonneuve P, Lowenfels AB. Risk factors for pancreatic cancer: a summary review of meta-analytical studies. Int J Epidemiol. 2014;44(1):186–98.
Article PubMed Google Scholar
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Al-Shaheri FN, Alhamdani MSS, Bauer AS, Giese N, Büchler MW, Hackert T, et al. Blood biomarkers for differential diagnosis and early detection of pancreatic cancer. Cancer Treat Rev. 2021;96: 102193.
Michl P, Löhr M, Neoptolemos JP, Capurso G, Rebours V, Malats N, et al. UEG position paper on pancreatic cancer. Bringing pancreatic cancer to the 21st century: Prevent, detect, and treat the disease earlier and better. United European Gastroenterol J. 2021;9(7):860–71.
Article PubMed PubMed Central Google Scholar
Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350(12):1200–10.
Chhoda A, Lu L, Clerkin BM, Risch H, Farrell JJ. Current approaches to pancreatic cancer screening. Am J Pathol. 2019;189(1):22–35.
Pantel K, Alix-Panabières C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med. 2010;16(9):398–406.
Kamyabi N, Bernard V, Maitra A. Liquid biopsies in pancreatic cancer. Expert Rev Anticancer Ther. 2019;19(10):869–78.
Article PubMed PubMed Central CAS Google Scholar
Lianidou E, Pantel K. Liquid biopsies. Genes Chromosomes Cancer. 2019;58(4):219–32.
Alix-Panabières C, Pantel K. Liquid biopsy: from discovery to clinical application. Cancer Discov. 2021;11(4):858–73.
Del Villano BC, Brennan S, Brock P, Bucher C, Liu V, McClure M, et al. Radioimmunometric assay for a monoclonal antibody-defined tumor marker, CA 19–9. Clin Chem. 1983;29(3):549–52.
Lee T, Teng TZJ, Shelat VG. Carbohydrate antigen 19–9 - tumor marker: Past, present, and future. World J Gastrointest Surg. 2020;12(12):468–90.
Goonetilleke KS, Siriwardena AK. Systematic review of carbohydrate antigen (CA 19–9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur J Surg Oncol. 2007;33(3):266–70.
Chan A, Prassas I, Dimitromanolakis A, Brand RE, Serra S, Diamandis EP, et al. Validation of biomarkers that complement CA19.9 in detecting early pancreatic cancer. Clin Cancer Res. 2014;20(22):5787–95.
Qu D, Johnson J, Chandrakesan P, Weygant N, May R, Aiello N, et al. Doublecortin-like kinase 1 is elevated serologically in pancreatic ductal adenocarcinoma and widely expressed on circulating tumor cells. PLoS ONE. 2015;10(2): e0118933.
Scarà S, Bottoni P, Scatena R. CA 19–9: biochemical and clinical aspects. Adv Exp Med Biol. 2015;867:247–60.
Alix-Panabières C, Pantel K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016;6(5):479–91.
Alix-Panabières C, Pantel K. Circulating Tumor Cells: Liquid Biopsy of Cancer. Clin Chem. 2013;59(1):110–8.
Nikanjam M, Kato S, Kurzrock R. Liquid biopsy: current technology and clinical applications. J Hematol Oncol. 2022;15(1):131.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Bingham C, Fernandez SV, Fittipaldi P, Dempsey PW, Ruth KJ, Cristofanilli M, et al. Mutational studies on single circulating tumor cells isolated from the blood of inflammatory breast cancer patients. Breast Cancer Res Treat. 2017;163(2):219–30.
Barradas AM, Terstappen LW. Towards the biological understanding of CTC: capture technologies, definitions and potential to create metastasis. Cancers (Basel). 2013;5(4):1619–42.
Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10(24):8152–62.
Smit DJ, Schneegans S, Pantel K. Clinical applications of circulating tumor cells in patients with solid tumors. Clin Exp Metastasis. 2024. https://doi.org/10.1007/s10585-024-10267-5 .
Wang Y, Yu X, Hartmann D, Zhou J. Circulating tumor cells in peripheral blood of pancreatic cancer patients and their prognostic role: a systematic review and meta-analysis. HPB (Oxford). 2020;22(5):660–9.
Zhu Y, Zhang H, Chen N, Hao J, Jin H, Ma X. Diagnostic value of various liquid biopsy methods for pancreatic cancer: a systematic review and meta-analysis. Medicine. 2020;99(3): e18581.
Komar G, Kauhanen S, Liukko K, Seppänen M, Kajander S, Ovaska J, et al. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin Cancer Res. 2009;15(17):5511–7.
Catenacci DV, Chapman CG, Xu P, Koons A, Konda VJ, Siddiqui UD, et al. Acquisition of portal venous circulating tumor cells from patients with pancreaticobiliary cancers by endoscopic ultrasound. Gastroenterology. 2015;149(7):1794–803.e4.
Markou A, Lazaridou M, Paraskevopoulos P, Chen S, Świerczewska M, Budna J, et al. Multiplex gene expression profiling of in vivo isolated circulating tumor cells in high-risk prostate cancer patients. Clin Chem. 2018;64(2):297–306.
Fehm TN, Meier-Stiegen F, Driemel C, Jäger B, Reinhardt F, Naskou J, et al. Diagnostic leukapheresis for CTC analysis in breast cancer patients: CTC frequency, clinical experiences and recommendations for standardized reporting. Cytometry A. 2018;93(12):1213–9.
Stoecklein NH, Fluegen G, Guglielmi R, Neves RPL, Hackert T, Birgin E, et al. Ultra-sensitive CTC-based liquid biopsy for pancreatic cancer enabled by large blood volume analysis. Mol Cancer. 2023;22(1):181.
Keller L, Pantel K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat Rev Cancer. 2019;19(10):553–67.
Khoja L, Backen A, Sloane R, Menasce L, Ryder D, Krebs M, et al. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br J Cancer. 2012;106(3):508–16.
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–61.
Ankeny JS, Court CM, Hou S, Li Q, Song M, Wu D, et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br J Cancer. 2016;114(12):1367–75.
Kuznetsova A, Popova O, Panchenkov D, Dyuzheva T, Ivanov A. Pancreatic ductal adenocarcinoma: tumor microenvironment and problems in the development of novel therapeutic strategies. Clin Exp Med. 2023;23(3):619–43.
Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer. 2019;18(1):14.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20.
Arneth B. Tumor Microenvironment. Medicina (Kaunas). 2019;56(1):15.
Götze J, Nitschke C, Uzunoglu FG, Pantel K, Sinn M, Wikman H. Tumor-stroma interaction in PDAC as a new approach for liquid biopsy and its potential clinical implications. Front Cell Dev Biol. 2022;10: 918795.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98.
Ao Z, Shah SH, Machlin LM, Parajuli R, Miller PC, Rawal S, et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015;75(22):4681–7.
Jones ML, Siddiqui J, Pienta KJ, Getzenberg RH. Circulating fibroblast-like cells in men with metastatic prostate cancer. Prostate. 2013;73(2):176–81.
Richardson AM, Havel LS, Koyen AE, Konen JM, Shupe J, Wiles WG IV, et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell–Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin Cancer Res. 2018;24(2):420–32.
Ortiz-Otero N, Marshall JR, Lash B, King MR. Chemotherapy-induced release of circulating-tumor cells into the bloodstream in collective migration units with cancer-associated fibroblasts in metastatic cancer patients. BMC Cancer. 2020;20(1):873.
Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–37.
Jaworski JJ, Morgan RD, Sivakumar S. Circulating cell-free tumour DNA for early detection of pancreatic cancer. Cancers (Basel). 2020;12(12):3704.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90.
Lo YMD, Zhang J, Leung TN, Lau TK, Chang AMZ, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64(1):218–24.
Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–9.
Keller L, Belloum Y, Wikman H, Pantel K. Clinical relevance of blood-based ctDNA analysis: mutation detection and beyond. Br J Cancer. 2021;124(2):345–58.
Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, et al. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov. 2015;5(10):1040–8.
Marin AM, Sanchuki HBS, Namur GN, Uno M, Zanette DL, Aoki MN. Circulating cell-free nucleic acids as biomarkers for diagnosis and prognosis of pancreatic cancer. Biomedicines. 2023;11(4):1069.
Jones S, Zhang X, Parsons DW, Lin JCH, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by Global Genomic Analyses. Science. 2008;321(5897):1801–6.
Saiki Y, Horii A. Molecular pathology of pancreatic cancer. Pathol Int. 2014;64(1):10–9.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Pelosi E, Castelli G, Testa U. Pancreatic cancer: molecular characterization, clonal evolution and cancer stem cells. Biomedicines. 2017;5(4):65.
Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016;538(7625):378–82.
Le Calvez-Kelm F, Foll M, Wozniak MB, Delhomme TM, Durand G, Chopard P, et al. KRAS mutations in blood circulating cell-free DNA: a pancreatic cancer case-control. Oncotarget. 2016;7(48):78827–40.
Conces M, Ni Y, Bazeley P, Patel B, Funchain P, Carraway HE. Clonal hematopoiesis of indeterminate potential (CHIP) mutations in solid tumor malignancies. J Clin Oncol. 2019;37(15):1507.
Article Google Scholar
Hoermann G. Clinical significance of clonal hematopoiesis of indeterminate potential in hematology and cardiovascular disease. Diagnostics (Basel). 2022;12(7):1613.
Qi T, Pan M, Shi H, Wang L, Bai Y, Ge Q. Cell-free DNA fragmentomics: the novel promising biomarker. Int J Mol Sci. 2023;24(2):1503.
Gorgannezhad L, Umer M, Islam MN, Nguyen N-T, Shiddiky MJA. Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip. 2018;18(8):1174–96.
Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–55.
Nicholson BD, Oke J, Virdee PS, Harris DA, O’Doherty C, Park JES, et al. Multi-cancer early detection test in symptomatic patients referred for cancer investigation in England and Wales (SYMPLIFY): a large-scale, observational cohort study. Lancet Oncol. 2023;24(7):733–43.
García-Ortiz MV, Cano-Ramírez P, Toledano-Fonseca M, Aranda E, Rodríguez-Ariza A. Diagnosing and monitoring pancreatic cancer through cell-free DNA methylation: progress and prospects. Biomark Res. 2023;11(1):88.
Xi H, Spencer CD, Peiyong J. Emerging frontiers of cell-free DNA fragmentomics. Extracellular Vesicles and Circulating Nucleic Acids. 2022;3(4):380–92.
Google Scholar
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164(1–2):57–68.
Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10(466):eaat4921.
Lo KW, Lo YM, Leung SF, Tsang YS, Chan LY, Johnson PJ, et al. Analysis of cell-free Epstein-Barr virus associated RNA in the plasma of patients with nasopharyngeal carcinoma. Clin Chem. 1999;45(8 Pt 1):1292–4.
Kopreski MS, Benko FA, Kwak LW, Gocke CD. Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clin Cancer Res. 1999;5(8):1961–5.
PubMed CAS Google Scholar
Heredia-Soto V, Rodríguez-Salas N, Feliu J. Liquid biopsy in pancreatic cancer: are we ready to apply it in the clinical practice? Cancers (Basel). 2021;13(8):1986.
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006.
Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–8.
Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE. 2008;3(11): e3694.
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011;108(12):5003–8.
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13(4):423–33.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Research. 2006;34(suppl_1):D140–4.
Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet. 2016;17(12):719–32.
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9:402.
van Rooij E. The art of microRNA research. Circ Res. 2011;108(2):219–34.
Cheung KWE, Choi SR, Lee LTC, Lee NLE, Tsang HF, Cheng YT, et al. The potential of circulating cell free RNA as a biomarker in cancer. Expert Rev Mol Diagn. 2019;19(7):579–90.
Peng C, Wang J, Gao W, Huang L, Liu Y, Li X, et al. Meta-analysis of the diagnostic performance of circulating MicroRNAs for pancreatic cancer. Int J Med Sci. 2021;18(3):660–71.
Schaaij-Visser TB, de Wit M, Lam SW, Jiménez CR. The cancer secretome, current status and opportunities in the lung, breast and colorectal cancer context. Biochim Biophys Acta. 2013;1834(11):2242–58.
Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–67.
Nagayoshi Y, Nakamura M, Matsuoka K, Ohtsuka T, Mori Y, Kono H, et al. Profiling of autoantibodies in sera of pancreatic cancer patients. Ann Surg Oncol. 2014;21(Suppl 3):S459–65.
Robinson JL, Feizi A, Uhlén M, Nielsen J. A systematic investigation of the malignant functions and diagnostic potential of the cancer secretome. Cell Rep. 2019;26(10):2622–35.e5.
Veyssière H, Bidet Y, Penault-Llorca F, Radosevic-Robin N, Durando X. Circulating proteins as predictive and prognostic biomarkers in breast cancer. Clin Proteomics. 2022;19(1):25.
Suhre K, McCarthy MI, Schwenk JM. Genetics meets proteomics: perspectives for large population-based studies. Nat Rev Genet. 2021;22(1):19–37.
O’Neill RS, Stoita A. Biomarkers in the diagnosis of pancreatic cancer: are we closer to finding the golden ticket? World J Gastroenterol. 2021;27(26):4045–87.
Kane LE, Mellotte GS, Mylod E, O’Brien RM, O’Connell F, Buckley CE, et al. Diagnostic accuracy of blood-based biomarkers for pancreatic cancer: a systematic review and meta-analysis. Cancer Res Commun. 2022;2(10):1229–43.
Fauré J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci. 2006;31(4):642–8.
Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36(13):1770–8.
Ge R, Tan E, Sharghi-Namini S, Asada HH. Exosomes in cancer microenvironment and beyond: have we overlooked these extracellular messengers? Cancer Microenvironment. 2012;5(3):323–32.
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9.
Whiteside TL. Tumor-derived exosomes and their role in cancer progression. Adv Clin Chem. 2016;74:103–41.
Armacki M, Polaschek S, Waldenmaier M, Morawe M, Ruhland C, Schmid R, et al. Protein kinase D1, reduced in human pancreatic tumors, increases secretion of small extracellular vesicles from cancer cells that promote metastasis to lung in mice. Gastroenterology. 2020;159(3):1019–35.e22.
Chang C-H, Pauklin S. Extracellular vesicles in pancreatic cancer progression and therapies. Cell Death Dis. 2021;12(11):973.
Ruivo CF, Bastos N, Adem B, Batista I, Duraes C, Melo CA, et al. Extracellular vesicles from pancreatic cancer stem cells lead an intratumor communication network (EVNet) to fuel tumour progression. Gut. 2022;71(10):2043–68.
Zhang YF, Zhou YZ, Zhang B, Huang SF, Li PP, He XM, et al. Pancreatic cancer-derived exosomes promoted pancreatic stellate cells recruitment by pancreatic cancer. J Cancer. 2019;10(18):4397–407.
Liu C, He D, Li L, Zhang S, Wang L, Fan Z, et al. Extracellular vesicles in pancreatic cancer immune escape: Emerging roles and mechanisms. Pharmacol Res. 2022;183: 106364.
Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177–82.
Patton MC, Zubair H, Khan MA, Singh S, Singh AP. Hypoxia alters the release and size distribution of extracellular vesicles in pancreatic cancer cells to support their adaptive survival. J Cell Biochem. 2020;121(1):828–39.
Jia E, Ren N, Shi X, Zhang R, Yu H, Yu F, et al. Extracellular vesicle biomarkers for pancreatic cancer diagnosis: a systematic review and meta-analysis. BMC Cancer. 2022;22(1):573.
Chang ST, Zahn JM, Horecka J, Kunz PL, Ford JM, Fisher GA, et al. Identification of a biomarker panel using a multiplex proximity ligation assay improves accuracy of pancreatic cancer diagnosis. J Transl Med. 2009;7:105.
Balasenthil S, Chen N, Lott ST, Chen J, Carter J, Grizzle WE, et al. A migration signature and plasma biomarker panel for pancreatic adenocarcinoma. Cancer Prev Res (Phila). 2011;4(1):137–49.
Brand RE, Nolen BM, Zeh HJ, Allen PJ, Eloubeidi MA, Goldberg M, et al. Serum biomarker panels for the detection of pancreatic cancer. Clin Cancer Res. 2011;17(4):805–16.
Park HD, Kang ES, Kim JW, Lee KT, Lee KH, Park YS, et al. Serum CA19-9, cathepsin D, and matrix metalloproteinase-7 as a diagnostic panel for pancreatic ductal adenocarcinoma. Proteomics. 2012;12(23–24):3590–7.
Nie S, Lo A, Wu J, Zhu J, Tan Z, Simeone DM, et al. Glycoprotein biomarker panel for pancreatic cancer discovered by quantitative proteomics analysis. J Proteome Res. 2014;13(4):1873–84.
Nolen BM, Brand RE, Prosser D, Velikokhatnaya L, Allen PJ, Zeh HJ, et al. Prediagnostic serum biomarkers as early detection tools for pancreatic cancer in a large prospective cohort study. PLoS ONE. 2014;9(4): e94928.
Shaw VE, Lane B, Jenkinson C, Cox T, Greenhalf W, Halloran CM, et al. Serum cytokine biomarker panels for discriminating pancreatic cancer from benign pancreatic disease. Mol Cancer. 2014;13:114.
Nie S, Yin H, Tan Z, Anderson MA, Ruffin MT, Simeone DM, et al. Quantitative analysis of single amino acid variant peptides associated with pancreatic cancer in serum by an isobaric labeling quantitative method. J Proteome Res. 2014;13(12):6058–66.
Tang H, Singh S, Partyka K, Kletter D, Hsueh P, Yadav J, et al. Glycan motif profiling reveals plasma sialyl-lewis x elevations in pancreatic cancers that are negative for sialyl-lewis A. Mol Cell Proteomics. 2015;14(5):1323–33.
Liu X, Zheng W, Wang W, Shen H, Liu L, Lou W, et al. A new panel of pancreatic cancer biomarkers discovered using a mass spectrometry-based pipeline. Br J Cancer. 2017;117(12):1846–54.
Mattila N, Seppänen H, Mustonen H, Przybyla B, Haglund C, Lassila R. Preoperative biomarker panel, including fibrinogen and FVIII, improves diagnostic accuracy for pancreatic ductal adenocarcinoma. Clin Appl Thromb Hemost. 2018;24(8):1267–75.
Peng HY, Chang MC, Hu CM, Yang HI, Lee WH, Chang YT. Thrombospondin-2 is a Highly Specific Diagnostic Marker and is Associated with Prognosis in Pancreatic Cancer. Ann Surg Oncol. 2019;26(3):807–14.
Staal B, Liu Y, Barnett D, Hsueh P, He Z, Gao C, et al. The sTRA Plasma biomarker: blinded validation of improved accuracy over CA19-9 in pancreatic cancer diagnosis. Clin Cancer Res. 2019;25(9):2745–54.
Zhou Q, Andersson R, Hu D, Bauden M, Sasor A, Bygott T, et al. Alpha-1-acid glycoprotein 1 is upregulated in pancreatic ductal adenocarcinoma and confers a poor prognosis. Transl Res. 2019;212:67–79.
Kim H, Kang KN, Shin YS, Byun Y, Han Y, Kwon W, et al. Biomarker panel for the diagnosis of pancreatic ductal adenocarcinoma. Cancers (Basel). 2020;12(6):1443. https://doi.org/10.3390/cancers12061443 .
Kim Y, Yeo I, Huh I, Kim J, Han D, Jang JY, et al. Development and multiple validation of the protein multi-marker panel for diagnosis of pancreatic cancer. Clin Cancer Res. 2021;27(8):2236–45.
Mehta S, Bhimani N, Gill AJ, Samra JS, Sahni S, Mittal A. Serum biomarker panel for diagnosis and prognosis of pancreatic ductal adenocarcinomas. Front Oncol. 2021;11: 708963.
Nené NR, Ney A, Nazarenko T, Blyuss O, Johnston HE, Whitwell HJ, et al. Serum biomarker-based early detection of pancreatic ductal adenocarcinomas with ensemble learning. Commun Med (Lond). 2023;3(1):10.
Cao H, Zhu L, Li L, Wang W, Niu X. Serum CA724 has no diagnostic value for gastrointestinal tumors. Clin Exp Med. 2023;23(6):2433–42.
Jang SI, Lee HK, Chang EJ, Kim S, Kim SY, Hong IY, et al. Improved predictability of pancreatic ductal adenocarcinoma diagnosis using a blood immune cell biomarker panel developed from bulk mRNA sequencing and single-cell RNA-sequencing. Cancer Immunol Immunother. 2023;72(8):2757–68.
Makawita S, Dimitromanolakis A, Soosaipillai A, Soleas I, Chan A, Gallinger S, et al. Validation of four candidate pancreatic cancer serological biomarkers that improve the performance of CA19.9. BMC Cancer. 2013;13:404.
Tang H, Partyka K, Hsueh P, Sinha JY, Kletter D, Zeh H, et al. Glycans related to the CA19-9 antigen are elevated in distinct subsets of pancreatic cancers and improve diagnostic accuracy over CA19-9. Cell Mol Gastroenterol Hepatol. 2016;2(2):201–21.e15.
PubMed Google Scholar
Yoneyama T, Ohtsuki S, Honda K, Kobayashi M, Iwasaki M, Uchida Y, et al. Identification of IGFBP2 and IGFBP3 as compensatory biomarkers for CA19-9 in early-stage pancreatic cancer using a combination of antibody-based and LC-MS/MS-based proteomics. PLoS ONE. 2016;11(8): e0161009.
Capello M, Bantis LE, Scelo G, Zhao Y, Li P, Dhillon DS, et al. Sequential validation of blood-based protein biomarker candidates for early-stage pancreatic cancer. J Natl Cancer Inst. 2017;109(4):djw266.
Balasenthil S, Huang Y, Liu S, Marsh T, Chen J, Stass SA, et al. A plasma biomarker panel to identify surgically resectable early-stage pancreatic cancer. J Natl Cancer Inst. 2017;109(8):djw341.
Kaur S, Smith LM, Patel A, Menning M, Watley DC, Malik SS, et al. A combination of MUC5AC and CA19-9 improves the diagnosis of pancreatic cancer: a multicenter study. Am J Gastroenterol. 2017;112(1):172–83.
Kim J, Bamlet WR, Oberg AL, Chaffee KG, Donahue G, Cao XJ, et al. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19–9 blood markers. Sci Transl Med. 2017;9(398):eaah5583.
Park J, Choi Y, Namkung J, Yi SG, Kim H, Yu J, et al. Diagnostic performance enhancement of pancreatic cancer using proteomic multimarker panel. Oncotarget. 2017;8(54):93117–30.
Resovi A, Bani MR, Porcu L, Anastasia A, Minoli L, Allavena P, et al. Soluble stroma-related biomarkers of pancreatic cancer. EMBO Mol Med. 2018;10(8):e8741.
Dong D, Jia L, Zhang L, Ma N, Zhang A, Zhou Y, et al. Periostin and CA242 as potential diagnostic serum biomarkers complementing CA19.9 in detecting pancreatic cancer. Cancer Sci. 2018;109(9):2841–51.
Song J, Sokoll LJ, Pasay JJ, Rubin AL, Li H, Bach DM, et al. Identification of serum biomarker panels for the early detection of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2019;28(1):174–82.
Jahan R, Ganguly K, Smith LM, Atri P, Carmicheal J, Sheinin Y, et al. Trefoil factor(s) and CA19.9: a promising panel for early detection of pancreatic cancer. EBioMedicine. 2019;42:375–85.
Zhang J, Wang Y, Zhao T, Li Y, Tian L, Zhao J, et al. Evaluation of serum MUC5AC in combination with CA19-9 for the diagnosis of pancreatic cancer. World J Surg Oncol. 2020;18(1):31.
Peng H, Pan S, Yan Y, Brand RE, Petersen GM, Chari ST, et al. Systemic proteome alterations linked to early stage pancreatic cancer in diabetic patients. Cancers (Basel). 2020;12(6):1534.
Choi YJ, Yoon W, Lee A, Han Y, Byun Y, Kang JS, et al. Diagnostic model for pancreatic cancer using a multi-biomarker panel. Ann Surg Treat Res. 2021;100(3):144–53.
Lee DH, Yoon W, Lee A, Han Y, Byun Y, Kang JS, et al. Multi-biomarker panel prediction model for diagnosis of pancreatic cancer. J Hepatobiliary Pancreat Sci. 2023;30(1):122–32.
Song J, Sokoll LJ, Chan DW, Zhang Z. Validation of serum biomarkers that complement CA19–9 in detecting early pancreatic cancer using electrochemiluminescent-based multiplex immunoassays. Biomedicines. 2021;9(12):1897.
Hinestrosa JP, Kurzrock R, Lewis JM, Schork NJ, Schroeder G, Kamat AM, et al. Early-stage multi-cancer detection using an extracellular vesicle protein-based blood test. Commun Med (Lond). 2022;2:29.
Firpo MA, Boucher KM, Bleicher J, Khanderao GD, Rosati A, Poruk KE, et al. Multianalyte serum biomarker panel for early detection of pancreatic adenocarcinoma. JCO Clin Cancer Inform. 2023;7: e2200160.
Byeon S, McKay MJ, Molloy MP, Gill AJ, Samra JS, Mittal A, et al. Novel serum protein biomarker panel for early diagnosis of pancreatic cancer. Int J Cancer. 2024;155:365–71.
Yi JM, Guzzetta AA, Bailey VJ, Downing SR, Van Neste L, Chiappinelli KB, et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin Cancer Res. 2013;19(23):6544–55.
Eissa MAL, Lerner L, Abdelfatah E, Shankar N, Canner JK, Hasan NM, et al. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin Epigenetics. 2019;11(1):59.
Ganepola GA, Rutledge JR, Suman P, Yiengpruksawan A, Chang DH. Novel blood-based microRNA biomarker panel for early diagnosis of pancreatic cancer. World J Gastrointest Oncol. 2014;6(1):22–33.
Makler A, Asghar W. Exosomal miRNA biomarker panel for pancreatic ductal adenocarcinoma detection in patient plasma: a pilot study. Int J Mol Sci. 2023;24(6):5081.
Yang Z, Huang J, Wu X, Zhou Y, Tang Y, Zhu Y, et al. Contribution of a circulating 2’-O-methylated MicroRNA panel to the diagnosis of pancreatic ductal adenocarcinoma. J Cancer. 2024;15(6):1583–92.
Madhavan B, Yue S, Galli U, Rana S, Gross W, Müller M, et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Cancer. 2015;136(11):2616–27.
Yuan W, Tang W, Xie Y, Wang S, Chen Y, Qi J, et al. New combined microRNA and protein plasmatic biomarker panel for pancreatic cancer. Oncotarget. 2016;7(48):80033–45.
Berger AW, Schwerdel D, Reinacher-Schick A, Uhl W, Algül H, Friess H, et al. A blood-based multi marker assay supports the differential diagnosis of early-stage pancreatic cancer. Theranostics. 2019;9(5):1280–7.
Reese M, Flammang I, Yang Z, Dhayat SA. Potential of exosomal microRNA-200b as liquid biopsy marker in pancreatic ductal adenocarcinoma. Cancers (Basel). 2020;12(1):197.
Chen J, Wang H, Zhou L, Liu Z, Tan X. A combination of circulating tumor cells and CA199 improves the diagnosis of pancreatic cancer. J Clin Lab Anal. 2022;36(5): e24341.
Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci U S A. 2017;114(38):10202–7.
Fahrmann JF, Bantis LE, Capello M, Scelo G, Dennison JB, Patel N, et al. A plasma-derived protein-metabolite multiplexed panel for early-stage pancreatic cancer. J Natl Cancer Inst. 2019;111(4):372–9.
Nakamura K, Zhu Z, Roy S, Jun E, Han H, Munoz RM, et al. An exosome-based transcriptomic signature for noninvasive, early detection of patients with pancreatic ductal adenocarcinoma: a multicenter cohort study. Gastroenterology. 2022;163(5):1252–66.e2.
Zhao R, Ren S, Li C, Guo K, Lu Z, Tian L, et al. Biomarkers for pancreatic cancer based on tissue and serum metabolomics analysis in a multicenter study. Cancer Med. 2023;12(4):5158–71.
Xu C, Jun E, Okugawa Y, Toiyama Y, Borazanci E, Bolton J, et al. A circulating panel of circRNA biomarkers for the noninvasive and early detection of pancreatic ductal adenocarcinoma. Gastroenterology. 2023;166(1)178–90.E16. https://doi.org/10.1053/j.gastro.2023.09.050 .
He J, Long J, Zhai C, Xu J, Bao K, Su W, et al. Codetection of proteins and RNAs on extracellular vesicles for pancreatic cancer early diagnosis. Anal Chem. 2024;96(17):6618–27.
Kim S, Park BK, Seo JH, Choi J, Choi JW, Lee CK, et al. Carbohydrate antigen 19–9 elevation without evidence of malignant or pancreatobiliary diseases. Sci Rep. 2020;10(1):8820.
Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Can Res. 2020;80(14):2969–74.
Article CAS Google Scholar
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–30.
Jamshidi A, Liu MC, Klein EA, Venn O, Hubbell E, Beausang JF, et al. Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell. 2022;40(12):1537–49.e12.
Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31(6):745–59.
Klein EA, Richards D, Cohn A, Tummala M, Lapham R, Cosgrove D, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol. 2021;32(9):1167–77.
Schrag D, Beer TM, McDonnell CH 3rd, Nadauld L, Dilaveri CA, Reid R, et al. Blood-based tests for multicancer early detection (PATHFINDER): a prospective cohort study. Lancet. 2023;402(10409):1251–60.
Gao Q, Lin YP, Li BS, Wang GQ, Dong LQ, Shen BY, et al. Unintrusive multi-cancer detection by circulating cell-free DNA methylation sequencing (THUNDER): development and independent validation studies. Ann Oncol. 2023;34(5):486–95.
Hartwig W, Hackert T, Hinz U, Gluth A, Bergmann F, Strobel O, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg. 2011;254(2):311–9.
Cameron JL, Riall TS, Coleman J, Belcher KA. One thousand consecutive pancreaticoduodenectomies. Ann Surg. 2006;244(1):10–5.
Yang R, Du Y, Zhang M, Liu Y, Feng H, Liu R, et al. Multi-omics analysis reveals interferon-stimulated gene OAS1 as a prognostic and immunological biomarker in pan-cancer. Front Immunol. 2023;14:1249731.
Lampignano R, Neumann MHD, Weber S, Kloten V, Herdean A, Voss T, et al. Multicenter evaluation of circulating cell-free DNA extraction and downstream analyses for the development of standardized (Pre)analytical work flows. Clin Chem. 2020;66(1):149–60.
Kloten V, Neumann MHD, Di Pasquale F, Sprenger-Haussels M, Shaffer JM, Schlumpberger M, et al. Multicenter evaluation of circulating plasma MicroRNA extraction technologies for the development of clinically feasible reverse transcription quantitative PCR and next-generation sequencing analytical work flows. Clin Chem. 2019;65(9):1132–40.
Neves RPL, Ammerlaan W, Andree KC, Bender S, Cayrefourcq L, Driemel C, et al. Proficiency Testing to Assess Technical Performance for CTC-Processing and Detection Methods in CANCER-ID. Clin Chem. 2021;67(4):631–41.
PANCAID Consortium. PANcreatic CAncer Initial Detection via liquid biopsy (PANCAID). 2023. Available from: https://pancaid-project.eu .
Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16(7):409–24.
Pascual J, Attard G, Bidard FC, Curigliano G, De Mattos-Arruda L, Diehn M, et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2022;33(8):750–68.
Pantel K, Lindbjerg Andersen C, Schuuring E, Malats N, Heitzer E, Vat L, et al. 237TiP GUIDE.MRD: a Consortium guiding multi-modal therapies against minimal residual disease (MRD) by liquid biopsy to assess implementation of circulating tumor DNA (ctDNA) in clinical practice to improve patient outcomes. Ann Oncol. 2023;34:S276–7.
Download references
Acknowledgements
This work was funded by the European Union. Views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or European Health and Digital Executive Agency (HADEA). Neither the European Union nor the granting authorities can be held responsible for them.
We would like to thank Sian Haynes-Ryterski for proofreading the manuscript.
Open Access funding enabled and organized by Projekt DEAL. K.P. received grant/research support from the EU/IHI GUIDE.MRD (No. 101112066), EU Horizon PANCAID (No. 101096309) and ERC Grant INJURMET (No. 834974). The position of K.-L.R. and in part the position of D.J.S. were funded through the PANCAID project within the EU Horizon Mission Cancer Program (No. 101096309)).
K.P. and D.J.S. received funding from the Fleur Hiege Center for Skin Cancer Research (funded by the Hiege Stiftung) outside of this work.
Author information
Authors and affiliations.
Institute of Tumor Biology, University Medical Center Hamburg-Eppendorf, Martinistraße 52, Hamburg, 20246, Germany
Kim-Lea Reese, Klaus Pantel & Daniel J. Smit
You can also search for this author in PubMed Google Scholar
Contributions
K.-L.R. reviewed the literature and wrote the original draft of the manuscript. K.P. and D.J.S. reviewed the literature and critically revised the original draft. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Correspondence to Klaus Pantel or Daniel J. Smit .
Ethics declarations
Ethics approval and consent to participate, consent for publication, competing interests.
K.P. received honoraria outside of this work from NRich, BMS, Agena, Menarini, Novartis, Sanofi, Illumina, Abcam, MSD, Eppendorf, and Hummingbird. K.-L.R. and D.J.S. declare that they have no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Reese, KL., Pantel, K. & Smit, D.J. Multibiomarker panels in liquid biopsy for early detection of pancreatic cancer – a comprehensive review. J Exp Clin Cancer Res 43 , 250 (2024). https://doi.org/10.1186/s13046-024-03166-w
Download citation
Received : 18 March 2024
Accepted : 16 August 2024
Published : 02 September 2024
DOI : https://doi.org/10.1186/s13046-024-03166-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Blood-based biomarkers
- Early-stage diagnosis
- Multimarker panel
- Pancreatic cancer
Journal of Experimental & Clinical Cancer Research
ISSN: 1756-9966
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 16 May 2024
A guide to artificial intelligence for cancer researchers
- Raquel Perez-Lopez ORCID: orcid.org/0000-0002-9176-0130 1 ,
- Narmin Ghaffari Laleh ORCID: orcid.org/0000-0003-0889-3352 2 ,
- Faisal Mahmood ORCID: orcid.org/0000-0001-7587-1562 3 , 4 , 5 , 6 , 7 , 8 &
- Jakob Nikolas Kather ORCID: orcid.org/0000-0002-3730-5348 2 , 9 , 10
Nature Reviews Cancer volume 24 , pages 427–441 ( 2024 ) Cite this article
15k Accesses
9 Citations
201 Altmetric
Metrics details
- Cancer imaging
- Mathematics and computing
- Tumour biomarkers
Artificial intelligence (AI) has been commoditized. It has evolved from a specialty resource to a readily accessible tool for cancer researchers. AI-based tools can boost research productivity in daily workflows, but can also extract hidden information from existing data, thereby enabling new scientific discoveries. Building a basic literacy in these tools is useful for every cancer researcher. Researchers with a traditional biological science focus can use AI-based tools through off-the-shelf software, whereas those who are more computationally inclined can develop their own AI-based software pipelines. In this article, we provide a practical guide for non-computational cancer researchers to understand how AI-based tools can benefit them. We convey general principles of AI for applications in image analysis, natural language processing and drug discovery. In addition, we give examples of how non-computational researchers can get started on the journey to productively use AI in their own work.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Artificial intelligence in histopathology: enhancing cancer research and clinical oncology

Guiding principles for the responsible development of artificial intelligence tools for healthcare

AI in health and medicine
Jiang, T., Gradus, J. L. & Rosellini, A. J. Supervised machine learning: a brief primer. Behav. Ther. 51 , 675–687 (2020).
Article PubMed PubMed Central Google Scholar
Alloghani, M., Al-Jumeily, D., Mustafina, J., Hussain, A. & Aljaaf, A. J. in Supervised and Unsupervised Learning for Data Science (eds Berry, M. W. et al.) 3–21 (Springer International, 2020).
Yala, A. et al. Optimizing risk-based breast cancer screening policies with reinforcement learning. Nat. Med. 28 , 136–143 (2022).
Article CAS PubMed Google Scholar
Kaufmann, E. et al. Champion-level drone racing using deep reinforcement learning. Nature 620 , 982–987 (2023).
Article CAS PubMed PubMed Central Google Scholar
Nasteski, V. An overview of the supervised machine learning methods. Horizons 4 , 51–62 (2017).
Article Google Scholar
Dike, H. U., Zhou, Y., Deveerasetty, K. K. & Wu, Q. Unsupervised learning based on artificial neural network: a review. In 2 018 IEEE International Conference on Cyborg and Bionic Systems (CBS) 322–327 (2018).
Shurrab, S. & Duwairi, R. Self-supervised learning methods and applications in medical imaging analysis: a survey. PeerJ Comput. Sci. 8 , e1045 (2022).
Wang, X. et al. Transformer-based unsupervised contrastive learning for histopathological image classification. Med. Image Anal. 81 , 102559 (2022).
Article PubMed Google Scholar
Wang, X. et al. RetCCL: clustering-guided contrastive learning for whole-slide image retrieval. Med. Image Anal. 83 , 102645 (2023).
Vinyals, O. et al. Grandmaster level in StarCraft II using multi-agent reinforcement learning. Nature 575 , 350–354 (2019).
Zhao, Y., Kosorok, M. R. & Zeng, D. Reinforcement learning design for cancer clinical trials. Stat. Med. 28 , 3294–3315 (2009).
Sapsford, R. & Jupp, V. Data Collection and Analysis (SAGE, 2006).
Yamashita, R., Nishio, M., Do, R. K. G. & Togashi, K. Convolutional neural networks: an overview and application in radiology. Insights Imaging 9 , 611–629 (2018).
Chowdhary, K. R. in Fundamentals of Artificial Intelligence (ed. Chowdhary, K. R.) 603–649 (Springer India, 2020).
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput. 9 , 1735–1780 (1997).
Vaswani, A. et al. Attention is all you need. Preprint at https://doi.org/10.48550/arXiv.1706.03762 (2017).
Shmatko, A., Ghaffari Laleh, N., Gerstung, M. & Kather, J. N. Artificial intelligence in histopathology: enhancing cancer research and clinical oncology. Nat. Cancer 3 , 1026–1038 (2022).
Wagner, S. J. et al. Transformer-based biomarker prediction from colorectal cancer histology: a large-scale multicentric study. Cancer Cell 41 , 1650–1661.e4 (2023).
Khan, A. et al. A survey of the vision transformers and their CNN-transformer based variants. Artif. Intell. Rev. 56 , 2917–2970 (2023).
Hamm, C. A. et al. Deep learning for liver tumor diagnosis part I: development of a convolutional neural network classifier for multi-phasic MRI. Eur. Radiol. 29 , 3338–3347 (2019).
Ren, J., Eriksen, J. G., Nijkamp, J. & Korreman, S. S. Comparing different CT, PET and MRI multi-modality image combinations for deep learning-based head and neck tumor segmentation. Acta Oncol. 60 , 1399–1406 (2021).
Unger, M. & Kather, J. N. A systematic analysis of deep learning in genomics and histopathology for precision oncology. BMC Med. Genomics 17 , 48 (2024).
Gawehn, E., Hiss, J. A. & Schneider, G. Deep learning in drug discovery. Mol. Inform. 35 , 3–14 (2016).
Bayramoglu, N., Kannala, J. & Heikkilä, J. Deep learning for magnification independent breast cancer histopathology image classification. In 2016 23rd International Conference on Pattern Recognition (ICPR) 2440–2445 (IEEE, 2016).
Galon, J. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313 , 1960–1964 (2006).
Schmidt, U., Weigert, M., Broaddus, C. & Myers, G. Cell detection with star-convex polygons. In Medical Image Computing and Computer Assisted Intervention — MICCAI 2018. Lecture Notes in Computer Science Vol. 11071 (eds Frangi, A. et al.) https://doi.org/10.1007/978-3-030-00934-2_30 (Springer, 2018).
Edlund, C. et al. LIVECell—a large-scale dataset for label-free live cell segmentation. Nat. Methods 18 , 1038–1045 (2021).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7 , 16878 (2017).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to imageJ: 25 years of image analysis. Nat. Methods 9 , 671–675 (2012).
Rueden, C. T. et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18 , 529 (2017).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9 , 676–682 (2012).
Linkert, M. et al. Metadata matters: access to image data in the real world. J. Cell Biol. 189 , 777–782 (2010).
Gómez-de-Mariscal, E. et al. DeepImageJ: a user-friendly environment to run deep learning models in ImageJ. Nat. Methods 18 , 1192–1195 (2021).
Betge, J. et al. The drug-induced phenotypic landscape of colorectal cancer organoids. Nat. Commun. 13 , 3135 (2022).
Park, T. et al. Development of a deep learning based image processing tool for enhanced organoid analysis. Sci. Rep. 13 , 19841 (2023).
Belthangady, C. & Royer, L. A. Applications, promises, and pitfalls of deep learning for fluorescence image reconstruction. Nat. Methods 16 , 1215–1225 (2019).
Echle, A. et al. Deep learning in cancer pathology: a new generation of clinical biomarkers. Br. J. Cancer 124 , 686–696 (2021).
Cifci, D., Foersch, S. & Kather, J. N. Artificial intelligence to identify genetic alterations in conventional histopathology. J. Pathol. 257 , 430–444 (2022).
Greenson, J. K. et al. Pathologic predictors of microsatellite instability in colorectal cancer. Am. J. Surg. Pathol. 33 , 126–133 (2009).
Kather, J. N. et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 25 , 1054–1056 (2019).
Echle, A. et al. Clinical-grade detection of microsatellite instability in colorectal tumors by deep learning. Gastroenterology 159 , 1406–1416.e11 (2020).
Kather, J. N. et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 1 , 789–799 (2020).
Fu, Y. et al. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat. Cancer 1 , 800–810 (2020).
Coudray, N. et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 24 , 1559–1567 (2018).
Schmauch, B. et al. A deep learning model to predict RNA-seq expression of tumours from whole slide images. Nat. Commun. 11 , 3877 (2020).
Binder, A. et al. Morphological and molecular breast cancer profiling through explainable machine learning. Nat. Mach. Intell. 3 , 355–366 (2021).
Loeffler, C. M. L. et al. Predicting mutational status of driver and suppressor genes directly from histopathology with deep learning: a systematic study across 23 solid tumor types. Front. Genet. 12 , 806386 (2022).
Chen, R. J. et al. Pan-cancer integrative histology-genomic analysis via multimodal deep learning. Cancer Cell 40 , 865–878.e6 (2022).
Bilal, M. et al. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: a retrospective study. Lancet Digit. Health 3 , e763–e772 (2021).
Yamashita, R. et al. Deep learning model for the prediction of microsatellite instability in colorectal cancer: a diagnostic study. Lancet Oncol. 22 , 132–141 (2021).
Echle, A. et al. Artificial intelligence for detection of microsatellite instability in colorectal cancer—a multicentric analysis of a pre-screening tool for clinical application. ESMO Open 7 , 100400 (2022).
Schirris, Y., Gavves, E., Nederlof, I., Horlings, H. M. & Teuwen, J. DeepSMILE: contrastive self-supervised pre-training benefits MSI and HRD classification directly from H&E whole-slide images in colorectal and breast cancer. Med. Image Anal. 79 , 102464 (2022).
Jain, M. S. & Massoud, T. F. Predicting tumour mutational burden from histopathological images using multiscale deep learning. Nat. Mach. Intell . 2 , 356–362 (2020).
Xu, H. et al. Spatial heterogeneity and organization of tumor mutation burden with immune infiltrates within tumors based on whole slide images correlated with patient survival in bladder cancer. J. Pathol. Inform. 13 , 100105 (2022).
Chen, S. et al. Deep learning-based approach to reveal tumor mutational burden status from whole slide images across multiple cancer types. Preprint at https://doi.org/10.48550/arXiv.2204.03257 (2023).
Shamai, G. et al. Artificial intelligence algorithms to assess hormonal status from tissue microarrays in patients with breast cancer. JAMA Netw. Open 2 , e197700 (2019).
Beck, A. H. et al. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci. Transl. Med. 3 , 108ra113 (2011).
Arslan, S. et al. A systematic pan-cancer study on deep learning-based prediction of multi-omic biomarkers from routine pathology images. Commun. Med. 4 , 48 (2024).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 25 , 1301–1309 (2019).
Lu, M. Y. et al. AI-based pathology predicts origins for cancers of unknown primary. Nature 594 , 106–110 (2021).
Kleppe, A. et al. A clinical decision support system optimising adjuvant chemotherapy for colorectal cancers by integrating deep learning and pathological staging markers: a development and validation study. Lancet Oncol. 23 , 1221–1232 (2022).
Jiang, X. et al. End-to-end prognostication in colorectal cancer by deep learning: a retrospective, multicentre study. Lancet Digit. Health 6 , e33–e43 (2024).
Zeng, Q. et al. Artificial intelligence-based pathology as a biomarker of sensitivity to atezolizumab–bevacizumab in patients with hepatocellular carcinoma: a multicentre retrospective study. Lancet Oncol. 24 , 1411–1422 (2023).
Ghaffari Laleh, N., Ligero, M., Perez-Lopez, R. & Kather, J. N. Facts and hopes on the use of artificial intelligence for predictive immunotherapy biomarkers in cancer. Clin. Cancer Res. 29 , 316–323 (2022).
Pedersen, A. et al. FastPathology: an open-source platform for deep learning-based research and decision support in digital pathology. IEEE Access 9 , 58216–58229 (2021).
Pocock, J. et al. TIAToolbox as an end-to-end library for advanced tissue image analytics. Commun. Med. 2 , 120 (2022).
Lu, M. Y. et al. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat. Biomed. Eng. 5 , 555–570 (2021).
El Nahhas, O. S. M. et al. From whole-slide image to biomarker prediction: a protocol for end-to-end deep learning in computational pathology. Preprint at https://doi.org/10.48550/arXiv.2312.10944 (2023).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. Preprint at https://doi.org/10.48550/arXiv.1912.01703 (2019).
Jorge Cardoso, M. et al. MONAI: an open-source framework for deep learning in healthcare. Preprint at https://doi.org/10.48550/arXiv.2211.02701 (2022).
Goode, A., Gilbert, B., Harkes, J., Jukic, D. & Satyanarayanan, M. OpenSlide: a vendor-neutral software foundation for digital pathology. J. Pathol. Inform. 4 , 27 (2013).
Martinez, K. & Cupitt, J. VIPS—a highly tuned image processing software architecture. In IEEE Int.Conf. Image Processing 2005 ; https://doi.org/10.1109/icip.2005.1530120 (2005).
Dolezal, J. M. et al. Deep learning generates synthetic cancer histology for explainability and education. NPJ Precis. Oncol. 7 , 49 (2023).
Plass, M. et al. Explainability and causability in digital pathology. Hip Int. 9 , 251–260 (2023).
Google Scholar
Reis-Filho, J. S. & Kather, J. N. Overcoming the challenges to implementation of artificial intelligence in pathology. J. Natl Cancer Inst. 115 , 608–612 (2023).
Aggarwal, R. et al. Diagnostic accuracy of deep learning in medical imaging: a systematic review and meta-analysis. NPJ Digit. Med. 4 , 65 (2021).
Rajput, D., Wang, W.-J. & Chen, C.-C. Evaluation of a decided sample size in machine learning applications. BMC Bioinformatics 24 , 48 (2023).
Ligero, M. et al. Minimizing acquisition-related radiomics variability by image resampling and batch effect correction to allow for large-scale data analysis. Eur. Radiol. 31 , 1460–1470 (2021).
Zwanenburg, A. et al. The image biomarker standardization initiative: standardized quantitative radiomics for high-throughput image-based phenotyping. Radiology 295 , 328–338 (2020).
van Griethuysen, J. J. M. et al. Computational radiomics system to decode the radiographic phenotype. Cancer Res. 77 , e104–e107 (2017).
Fedorov, A. et al. 3D Slicer as an image computing platform for the quantitative imaging network. Magn. Reson. Imaging 30 , 1323–1341 (2012).
Yushkevich, P. A. et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 31 , 1116–1128 (2006).
Khader, F. et al. Multimodal deep learning for integrating chest radiographs and clinical parameters: a case for transformers. Radiology 309 , e230806 (2023).
Yu, A. C., Mohajer, B. & Eng, J. External validation of deep learning algorithms for radiologic diagnosis: a systematic review. Radiol. Artif. Intell. 4 , e210064 (2022).
US FDA. Artificial intelligence and machine learning (AI/ML)-enabled medical devices; https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-aiml-enabled-medical-devices (2023).
Bruker Corporation. Artificial intelligence in NMR; https://www.bruker.com/en/landingpages/bbio/artificial-intelligence-in-nmr.html (2024).
Wasserthal, J. TotalSegmentator: tool for robust segmentation of 104 important anatomical structures in CT images. GitHub https://doi.org/10.5281/zenodo.6802613 (2023).
Garcia-Ruiz, A. et al. An accessible deep learning tool for voxel-wise classification of brain malignancies from perfusion MRI. Cell Rep. Med. 5 , 101464 (2024).
Lång, K. et al. Artificial intelligence-supported screen reading versus standard double reading in the Mammography Screening with Artificial Intelligence trial (MASAI): a clinical safety analysis of a randomised, controlled, non-inferiority, single-blinded, screening accuracy study. Lancet Oncol. 24 , 936–944 (2023).
Bera, K., Braman, N., Gupta, A., Velcheti, V. & Madabhushi, A. Predicting cancer outcomes with radiomics and artificial intelligence in radiology. Nat. Rev. Clin. Oncol. 19 , 132–146 (2022).
Núñez, L. M. et al. Unraveling response to temozolomide in preclinical GL261 glioblastoma with MRI/MRSI using radiomics and signal source extraction. Sci. Rep. 10 , 19699 (2020).
Müller, J. et al. Radiomics-based tumor phenotype determination based on medical imaging and tumor microenvironment in a preclinical setting. Radiother. Oncol. 169 , 96–104 (2022).
Amirrashedi, M. et al. Leveraging deep neural networks to improve numerical and perceptual image quality in low-dose preclinical PET imaging. Comput. Med. Imaging Graph. 94 , 102010 (2021).
Zinn, P. O. et al. A coclinical radiogenomic validation study: conserved magnetic resonance radiomic appearance of periostin-expressing glioblastoma in patients and xenograft models. Clin. Cancer Res. 24 , 6288–6299 (2018).
Lin, Y.-C. et al. Diffusion radiomics analysis of intratumoral heterogeneity in a murine prostate cancer model following radiotherapy: pixelwise correlation with histology. J. Magn. Reson. Imaging 46 , 483–489 (2017).
Moor, M. et al. Foundation models for generalist medical artificial intelligence. Nature 616 , 259–265 (2023).
Chen, R. J. et al. Towards a general-purpose foundation model for computational pathology. Nat. Med. 30 , 850–862 (2024).
Unger, M. & Kather, J. N. Deep learning in cancer genomics and histopathology. Genome Med. 16 , 44 (2024).
Zhou, Y. et al. A foundation model for generalizable disease detection from retinal images. Nature 622 , 156–163 (2023).
Filiot, A. et al. Scaling self-supervised learning for histopathology with masked image modeling. Preprint at bioRxiv https://doi.org/10.1101/2023.07.21.23292757 (2023).
Campanella, G. et al. Computational pathology at health system scale—self-supervised foundation models from three billion images. Preprint at https://doi.org/10.48550/arXiv.2310.07033 (2023).
Vorontsov, E. et al. Virchow: a million-slide digital pathology foundation model. Preprint at https://doi.org/10.48550/arXiv.2309.07778 (2023).
Clusmann, J. et al. The future landscape of large language models in medicine. Commun. Med. 3 , 141 (2023).
Bubeck, S. et al. Sparks of artificial general intelligence: early experiments with GPT-4. Preprint at https://doi.org/10.48550/arXiv.2303.12712 (2023).
Truhn, D., Reis-Filho, J. S. & Kather, J. N. Large language models should be used as scientific reasoning engines, not knowledge databases. Nat. Med. 29 , 2983–2984 (2023).
Adams, L. C. et al. Leveraging GPT-4 for post hoc transformation of free-text radiology reports into structured reporting: a multilingual feasibility study. Radiology 307 , e230725 (2023).
Truhn, D. et al. Extracting structured information from unstructured histopathology reports using generative pre-trained transformer 4 (GPT-4). J. Pathol. 262 , 310–319 (2023).
Wiest, I. C. et al. From text to tables: a local privacy preserving large language model for structured information retrieval from medical documents. Preprint at bioRxiv https://doi.org/10.1101/2023.12.07.23299648 (2023).
Singhal, K. et al. Large language models encode clinical knowledge. Nature 620 , 172–180 (2023).
Truhn, D. et al. A pilot study on the efficacy of GPT-4 in providing orthopedic treatment recommendations from MRI reports. Sci. Rep. 13 , 20159 (2023).
Wang, H. et al. Scientific discovery in the age of artificial intelligence. Nature 620 , 47–60 (2023).
Derraz, B. et al. New regulatory thinking is needed for AI-based personalised drug and cell therapies in precision oncology. NPJ Precis. Oncol . https://doi.org/10.1038/s41698-024-00517-w (2024).
Extance, A. ChatGPT has entered the classroom: how LLMs could transform education. Nature 623 , 474–477 (2023).
Thirunavukarasu, A. J. et al. Large language models in medicine. Nat. Med. 29 , 1930–1940 (2023).
Webster, P. Six ways large language models are changing healthcare. Nat. Med. 29 , 2969–2971 (2023).
Krishnan, R., Rajpurkar, P. & Topol, E. J. Self-supervised learning in medicine and healthcare. Nat. Biomed. Eng. 6 , 1346–1352 (2022).
Meskó, B. Prompt engineering as an important emerging skill for medical professionals: tutorial. J. Med. Internet Res. 25 , e50638 (2023).
Sushil, M. et al. CORAL: expert-curated oncology reports to advance language model inference. NEJM AI 1 , 4 (2024).
Brown, T. B. et al. Language models are few-shot learners. Preprint at https://doi.org/10.48550/arXiv.2005.01416 (2020).
Ferber, D. & Kather, J. N. Large language models in uro-oncology. Eur. Urol. Oncol. 7 , 157–159 (2023).
Jiang, L. Y. et al. Health system-scale language models are all-purpose prediction engines. Nature 619 , 357–362 (2023).
Nori, H. et al. Can generalist foundation models outcompete special-purpose tuning? Case study in medicine. Preprint at https://doi.org/10.48550/arXiv.2311.16452 (2023).
Balaguer, A. et al. RAG vs fine-tuning: pipelines, tradeoffs, and a case study on agriculture. Preprint at https://doi.org/10.48550/arXiv.2401.08406 (2024).
Gemini Team et al. Gemini: a family of highly capable multimodal models. Preprint at https://doi.org/10.48550/arXiv.2312.11805 (2023).
Tisman, G. & Seetharam, R. OpenAI’s ChatGPT-4, BARD and YOU.Com (AI) and the cancer patient, for now, caveat emptor, but stay tuned. Digit. Med. Healthc. Technol. https://doi.org/10.5772/dmht.19 (2023).
Touvron, H. et al. LLaMA: open and efficient foundation language models. Preprint at https://doi.org/10.48550/arXiv.2302.13971 (2023).
Lipkova, J. et al. Artificial intelligence for multimodal data integration in oncology. Cancer Cell 40 , 1095–1110 (2022).
Niehues, J. M. et al. Generalizable biomarker prediction from cancer pathology slides with self-supervised deep learning: a retrospective multi-centric study. Cell Rep. Med. 4 , 100980 (2023).
Foersch, S. et al. Multistain deep learning for prediction of prognosis and therapy response in colorectal cancer. Nat. Med. 29 , 430–439 (2023).
Boehm, K. M. et al. Multimodal data integration using machine learning improves risk stratification of high-grade serous ovarian cancer. Nat. Cancer 3 , 723–733 (2022).
Vanguri, R. et al. Multimodal integration of radiology, pathology and genomics for prediction of response to PD-(L)1 blockade in patients with non-small cell lung cancer. Nat. Cancer 3 , 1151–1164 (2022).
Shifai, N., van Doorn, R., Malvehy, J. & Sangers, T. E. Can ChatGPT vision diagnose melanoma? An exploratory diagnostic accuracy study. J. Am. Acad. Dermatol . 90 , 1057–1059 (2024).
Liu, H., Li, C., Wu, Q. & Lee, Y. J. Visual instruction tuning. Preprint at https://doi.org/10.48550/arXiv.2304.08485 (2023).
Li, C. et al. LLaVA-med: training a large language-and-vision assistant for biomedicine in one day. Preprint at https://doi.org/10.48550/arXiv.2306.00890 (2023).
Lu, M. Y. et al. A foundational multimodal vision language AI assistant for human pathology. Preprint at https://doi.org/10.48550/arXiv.2312.07814 (2023).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8 , 1324 (2017).
Zhang, Z. et al. Uniform genomic data analysis in the NCI Genomic Data Commons. Nat. Commun. 12 , 1226 (2021).
Vega, D. M. et al. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: phase II of the Friends of Cancer Research TMB Harmonization Project. Ann. Oncol. 32 , 1626–1636 (2021).
Anaya, J., Sidhom, J.-W., Mahmood, F. & Baras, A. S. Multiple-instance learning of somatic mutations for the classification of tumour type and the prediction of microsatellite status. Nat. Biomed. Eng. 8 , 57–67 (2023).
Chen, B. et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 37 , 1332–1343 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596 , 583–589 (2021).
Callaway, E. What’s next for AlphaFold and the AI protein-folding revolution. Nature 604 , 234–238 (2022).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381 , eadg7492 (2023).
Barrio-Hernandez, I. et al. Clustering predicted structures at the scale of the known protein universe. Nature 622 , 637–645 (2023).
Yang, X., Wang, Y., Byrne, R., Schneider, G. & Yang, S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 119 , 10520–10594 (2019).
Mullowney, M. W. et al. Artificial intelligence for natural product drug discovery. Nat. Rev. Drug Discov. 22 , 895–916 (2023).
Jayatunga, M. K. P., Xie, W., Ruder, L., Schulze, U. & Meier, C. AI in small-molecule drug discovery: a coming wave? Nat. Rev. Drug Discov. 21 , 175–176 (2022).
Vert, J.-P. How will generative AI disrupt data science in drug discovery? Nat. Biotechnol. 41 , 750–751 (2023).
Wong, F. et al. Discovery of a structural class of antibiotics with explainable deep learning. Nature 626 , 177–185 (2023).
Swanson, K. et al. Generative AI for designing and validating easily synthesizable and structurally novel antibiotics. Nat. Mach. Intell. 6 , 338–353 (2024).
Janizek, J. D. et al. Uncovering expression signatures of synergistic drug responses via ensembles of explainable machine-learning models. Nat. Biomed. Eng. 7 , 811–829 (2023).
Savage, N. Drug discovery companies are customizing ChatGPT: here’s how. Nat. Biotechnol. 41 , 585–586 (2023).
Boiko, D. A., MacKnight, R., Kline, B. & Gomes, G. Autonomous chemical research with large language models. Nature 624 , 570–578 (2023).
Arnold, C. AlphaFold touted as next big thing for drug discovery—but is it? Nature 622 , 15–17 (2023).
Mock, M., Edavettal, S., Langmead, C. & Russell, A. AI can help to speed up drug discovery—but only if we give it the right data. Nature 621 , 467–470 (2023).
AI’s potential to accelerate drug discovery needs a reality check. Nature 622 , 217 (2023).
Upswing in AI drug-discovery deals. Nat. Biotechnol . 41 , 1361 (2023).
Hutson, M. AI for drug discovery is booming, but who owns the patents? Nat. Biotechnol. 41 , 1494–1496 (2023).
Wong, C. H., Siah, K. W. & Lo, A. W. Estimation of clinical trial success rates and related parameters. Biostatistics 20 , 273–286 (2019).
Subbiah, V. The next generation of evidence-based medicine. Nat. Med. 29 , 49–58 (2023).
Yuan, C. et al. Criteria2Query: a natural language interface to clinical databases for cohort definition. J. Am. Med. Inform. Assoc. 26 , 294–305 (2019).
Lu, L., Dercle, L., Zhao, B. & Schwartz, L. H. Deep learning for the prediction of early on-treatment response in metastatic colorectal cancer from serial medical imaging. Nat. Commun. 12 , 6654 (2021).
Trebeschi, S. et al. Prognostic value of deep learning-mediated treatment monitoring in lung cancer patients receiving immunotherapy. Front. Oncol. 11 , 609054 (2021).
Castelo-Branco, L. et al. ESMO guidance for reporting oncology real-world evidence (GROW). Ann. Oncol. 34 , 1097–1112 (2023).
Morin, O. et al. An artificial intelligence framework integrating longitudinal electronic health records with real-world data enables continuous pan-cancer prognostication. Nat. Cancer 2 , 709–722 (2021).
Yang, X. et al. A large language model for electronic health records. NPJ Digit. Med. 5 , 194 (2022).
Huang, X., Rymbekova, A., Dolgova, O., Lao, O. & Kuhlwilm, M. Harnessing deep learning for population genetic inference. Nat. Rev. Genet. 25 , 61–78 (2024).
Pawlicki, Lee, D.-S., Hull & Srihari. Neural network models and their application to handwritten digit recognition. In IEEE 1988 Int. Conf. Neural Networks (eds Pawlicki, T. F. et al.) 63–70 (1988).
Chui, M. et al. The economic potential of generative AI: the next productivity frontier. McKinsey https://www.mckinsey.com/capabilities/mckinsey-digital/our-insights/the-economic-potential-of-generative-ai-the-next-productivity-frontier (2023).
Dell’Acqua, F. et al. Navigating the jagged technological frontier: field experimental evidence of the effects of AI on knowledge worker productivity and quality. Harvard Business School https://www.hbs.edu/ris/Publication%20Files/24-013_d9b45b68-9e74-42d6-a1c6-c72fb70c7282.pdf (2023).
Boehm, K. M., Khosravi, P., Vanguri, R., Gao, J. & Shah, S. P. Harnessing multimodal data integration to advance precision oncology. Nat. Rev. Cancer 22 , 114–126 (2022).
Gilbert, S., Harvey, H., Melvin, T., Vollebregt, E. & Wicks, P. Large language model AI chatbots require approval as medical devices. Nat. Med. 29 , 2396–2398 (2023).
Mobadersany, P. et al. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc. Natl Acad. Sci. USA 115 , E2970–E2979 (2018).
Chang, Y. et al. A survey on evaluation of large language models. ACM Trans. Intell. Syst. Technol. 15 , 1–45 (2024).
Lin, T., Wang, Y., Liu, X. & Qiu, X. A survey of transformers. AI Open 3 , 111–132 (2022).
Download references
Acknowledgements
R.P.-L. is supported by LaCaixa Foundation, a CRIS Foundation Talent Award (TALENT19-05), the FERO Foundation, the Instituto de Salud Carlos III-Investigacion en Salud (PI18/01395 and PI21/01019), the Prostate Cancer Foundation (18YOUN19) and the Asociación Española Contra el Cancer (AECC) (PRYCO211023SERR). J.N.K. is supported by the German Cancer Aid (DECADE, 70115166), the German Federal Ministry of Education and Research (PEARL, 01KD2104C; CAMINO, 01EO2101; SWAG, 01KD2215A; TRANSFORM LIVER, 031L0312A; and TANGERINE, 01KT2302 through ERA-NET Transcan), the German Academic Exchange Service (SECAI, 57616814), the German Federal Joint Committee (TransplantKI, 01VSF21048), the European Union’s Horizon Europe and innovation programme (ODELIA, 101057091; and GENIAL, 101096312), the European Research Council (ERC; NADIR, 101114631) and the National Institute for Health and Care Research (NIHR; NIHR203331) Leeds Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. This work was funded by the European Union. Views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union. Neither the European Union nor the granting authority can be held responsible for them.
Author information
Authors and affiliations.
Radiomics Group, Vall d’Hebron Institute of Oncology, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain
Raquel Perez-Lopez
Else Kroener Fresenius Center for Digital Health, Technical University Dresden, Dresden, Germany
Narmin Ghaffari Laleh & Jakob Nikolas Kather
Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
Faisal Mahmood
Department of Pathology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Cancer Program, Broad Institute of Harvard and MIT, Cambridge, MA, USA
Cancer Data Science Program, Dana-Farber Cancer Institute, Boston, MA, USA
Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA
Harvard Data Science Initiative, Harvard University, Cambridge, MA, USA
Department of Medicine I, University Hospital Dresden, Dresden, Germany
Jakob Nikolas Kather
Medical Oncology, National Center for Tumour Diseases (NCT), University Hospital Heidelberg, Heidelberg, Germany
You can also search for this author in PubMed Google Scholar
Contributions
All authors contributed substantially to discussion of the content and reviewed and/or edited the manuscript before the submission. R.P.-L., N.G.L. and J.N.K. researched data for the article and wrote the article.
Corresponding author
Correspondence to Jakob Nikolas Kather .
Ethics declarations
Competing interests.
J.N.K. declares consulting services for Owkin, DoMore Diagnostics, Panakeia, Scailyte, Mindpeak and MultiplexDx; holds shares in StratifAI GmbH; has received a research grant from GSK; and has received honoraria from AstraZeneca, Bayer, Eisai, Janssen, MSD, BMS, Roche, Pfizer and Fresenius. R.P.-L. declares research funding by AstraZeneca and Roche, and participates in the steering committee of a clinical trial sponsored by Roche, not related to this work. All other authors declare no competing interests.
Peer review
Peer review information.
Nature Reviews Cancer thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Hugging Face: https://huggingface.co/
You.com: https://you.com
Supplementary information
Supplementary information.
(API). A set of tools and protocols for building software and applications, enabling software to communicate with AI models.
(ANNs). Computational models loosely inspired by the structure and function of the human brain, consisting of interconnected layers of nodes, called neurons, that process input data and learn to recognize patterns and make decisions.
The use of algorithms, machine learning and image analysis techniques to extract information from digital pathology images.
A field of AI that focuses on enabling computers to analyse and interpret visual data, such as images and videos.
(CNNs). A type of deep neural network that is especially effective for analysing visual imagery and used in image analysis.
Deep learning is a subfield of machine learning that uses artificial neural networks with multiple layers, called deep neural networks, to learn and extract highly complex features and patterns from raw input data.
Visual representations captured and stored in a digital format, consisting of a grid of pixels, with each pixel representing a colour intensity value.
The practice of converting glass slides into digital slides that can be viewed, managed and analysed on a computer.
Techniques in AI that provide insights and explanations on how the AI model arrived at its conclusions, thus making the decision-making process of the AI more transparent.
AI systems that can generate new content (text, images or music) that is similar to the content on which it was trained, often creating novel and coherent outputs.
Extremely high-resolution digital images consisting of 1 billion pixels, obtained by scanning tissue slides with a slide scanner.
(GPUs). Specialized hardware used to rapidly process large blocks of data simultaneously, used in computer gaming and AI.
(LLMs). Advanced AI models trained on vast amounts of text data, capable of analysing, generating and manipulating human language, often at the human level 174 .
A type of neural network particularly good at processing sequences of data (such as time series or language), with a capability to remember information for a certain time.
A subset of AI focusing on the development of algorithms and models that enable computers to learn and improve their performance on a specific task without being explicitly instructed how to achieve this.
(NLP). A branch of AI that helps computers to analyse, interpret and respond to human language in a useful way.
Crafting inputs or questions in a way that guides AI models, particularly LLMs, to provide the most effective and accurate responses.
Types of a neural network model that excel at processing sequences of data, such as sentences in text, by focusing on different parts of the sequence to make predictions 175 .
The three-dimensional equivalent of a pixel in images, representing a value on a regular grid in three-dimensional space, commonly used in medical imaging such as MRI and CT scans.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Cite this article.
Perez-Lopez, R., Ghaffari Laleh, N., Mahmood, F. et al. A guide to artificial intelligence for cancer researchers. Nat Rev Cancer 24 , 427–441 (2024). https://doi.org/10.1038/s41568-024-00694-7
Download citation
Accepted : 09 April 2024
Published : 16 May 2024
Issue Date : June 2024
DOI : https://doi.org/10.1038/s41568-024-00694-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Advancements in triple-negative breast cancer sub-typing, diagnosis and treatment with assistance of artificial intelligence : a focused review.
- Zahra Batool
- Mohammad Amjad Kamal
- Bairong Shen
Journal of Cancer Research and Clinical Oncology (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Cancer newsletter — what matters in cancer research, free to your inbox weekly.


An official website of the United States government
Here’s how you know
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( Lock A locked padlock ) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
https://www.nist.gov/news-events/news/2024/09/spotlight-game-changing-microscopy-technique-identifying-cancerous-cells
Spotlight: Game-Changing Microscopy Technique for Identifying Cancerous Cells

Breast-conserving surgery, or lumpectomy, is a treatment for early-stage breast cancer. It’s important for surgeons to remove enough tissue around the tumor to ensure that no cancer cells are left, but still conserve the surrounding healthy tissue as much as possible. Traditionally, this requires a slow process of staining and examining tissue under a microscope.
Researchers at NIST have developed a faster imaging method using hyperspectral dark-field microscopy. This technique uses different wavelengths of light to distinguish between cancerous and healthy cells without staining. It can be used on fresh tissue samples during surgery, providing immediate feedback to the surgeon. This reduces the need for follow-up surgeries by ensuring that all cancerous tissue is removed in the first operation.
This new method, combined with machine learning, accurately identifies cancer cells and could greatly improve breast cancer treatment and patient care.
As breast cancer remains a leading cause of cancer-related deaths in women, innovations like these are crucial in improving treatment and overall patient care.
Learn more about the method.
Follow us on social media for more like this from all across NIST!
- Become a member

Official Media

Young’s Seafood adds Taste of India range to Gastro brand of products
Ólafur karl sigurdarson leaves marel for kapp; håkon andré berg named executive chairman at geosalmo, ode's farmed cod featured at world sushi cup, indian shrimp added to us list of goods likely made with forced labor, kingspan marketing its quadcore insulation to asian seafood businesses, biopsy techniques used in human cancer research showing promise in mussel testing, dylan sloan.
- News » Food Safety & Health
Canadian researchers applying oncology techniques to the study of mussel species have claimed their research provides a way for scientists to comprehensively test for marine pathogens in the bivalves that’s simpler, cheaper, and faster than current alternatives.
“Our approach has the potential to detect all the viruses, all the bacteria, all the pathogens,” Professor Yves St-Pierre of Canada’s Institut national de la recherche scientifique in Quebec City, Montreal, told SeafoodSource. “It's a more integrated approach that allows us to get so much more information than what [we] can get right now.”
Like other bivalves, mussels filter lots of water – up to 15 gallons per day . The volume of water with which they come into contact means that mussels filter and retain relatively high quantities of the pathogens in their ecosystem, soaking up many bacteria, viruses, and other organisms that serve as indicators of marine heath in a particular area, also known as “ sentinel species .”
However, comprehensively sampling and interpreting DNA from the full range of trace pathogens stored in mussels has proven challenging for researchers.
That’s where St-Pierre comes in.
St-Pierre has been a cancer researcher for over 20 years and first experimented with the approach of applying cancer-detection techniques to marine life a decade ago on a visit to the Kerguelen Islands, a remote archipelago over 1,000 miles off the coast of Antarctica. Now, a Quebec-based team led by St-Pierre and doctoral student Sophia Ferchiou has released a paper showing evidence that cancer-detection techniques could have strong use cases for pathogen detection in the aquaculture industry and for marine environmental monitoring.
The researchers utilized a technique called liquid biopsy, commonly used in cancer treatment, to test mussels at four sites in Quebec. In oncology, doctors use liquid biopsy to detect small blood samples of cancer-causing DNA in a person’s bloodstream. Applying that same method to mussels and other invertebrates allowed the researchers to isolate and identify an array of pathogens, bacteria, and other environmental indicators that were present in the ecosystem and stored in the mussels.
St-Pierre was surprised at the sheer amount of data he and the team were able to extract from individual samples, which included the presence and relative levels of harmful marine pathogens present in the mussels’ ecosystem, mutations, and genetic changes in the mussel population, as well as the presence of certain fish species in the area.
“We even discovered viruses … that were affecting [nearby] terrestrial organisms, like cattle. Because if it rains, you have DNA running into water,” St-Pierre said. “A mussel picks up the DNA, we sequence it, and then we can find a virus or a pathogen that is in a nearby ecosystem.”
Better yet for the possibility of future research expansion, recent advances in DNA sequencing technology have made liquid biopsy techniques the researchers used far cheaper and easier …

Become a Premium member to unlock the rest of this article.
Continue reading ›
Already a member? Log in ›
Want seafood news sent to your inbox?

Cancer Center researcher studies combination therapy to improve leukemia treatment
Lls, asco grants fund early career physician-scientist.

The University of Cincinnati Cancer Center’s Eric Vick, MD, PhD, has been awarded a nearly $215,000 Career Development Award from the Leukemia and Lymphoma Society (LLS) and a $50,000 American Society of Clinical Oncology (ASCO) Young Investigator Award to continue research into a combination therapy treatment for acute myeloid leukemia (AML).
Eric Vick, MD, PhD. Photo/Andrew Higley/UC Marketing + Brand.
A protein called IRAK4 is known to help drive AML cell growth through overactivation, but targeting this protein alone has not proven to be an effective treatment for killing the cancer cells. Research Vick presented at the American Society of Hematology annual meeting last December looked at potential compounds to pair with drugs that target IRAK4 as a more effective combination therapy.
“After you have combined the loss of IRAK4 with different pharmacologic agents, you create a situation that is lethal for the cells,” said Vick, instructor in the Division of Hematology/Oncology in UC’s College of Medicine and a UC Health attending physician at the Cancer Center’s Blood Cancer Healing Center.
After testing more than 2,800 small molecules to compare their effectiveness in killing both normal AML cells and those that did not have overactive IRAK4, Vick and colleagues found one compound was especially effective at killing AML cells by reducing the levels of a different protein, called c-Myc, that also drives cancer growth.
“We see that IRAK4 inhibition or deletion has a unique role in decreasing the lifespan of c-Myc. By combining it with different compounds, we can decrease the amount of c-Myc and, by doing so, decrease cell growth in leukemia,” Vick said. “A major focus of this grant will be understanding how IRAK4 inhibition decreases c-Myc.”
The three-year LLS grant will allow Vick to further his research and describe in more detail exactly how targeting IRAK4 affects c-Myc. The one-year ASCO grant will increase the team’s bandwidth and include the study of additional potential compounds to use in the combination treatment.
“c-Myc has been described as untargetable, and there are many groups who have tried to target this protein without clinical success,” Vick said. “Our hope is that by understanding how IRAK4 targets c-Myc, we can indirectly lead to a loss of c-Myc. This may be the only way to target something ubiquitous and essential to normal cellular processes, but at the core of so many malignancies.”
For Dr. Vick, this award is a testament to the impact he has already made in leukemia research and the promise he holds as a physician-scientist.
Daniel Starczynowski, PhD
With this additional knowledge, the ultimate aim of this grant funding is to move the research from observation in the lab to intervention in the clinic through a trial testing combination therapies targeting IRAK4 and c-Myc. Vick and colleagues will also investigate other diseases with similar protein mechanisms that could benefit from the same proposed treatment.
Vick worked as a clinical fellow in Daniel Starczynowski’s lab at Cincinnati Children’s Hospital prior to joining UC’s faculty, and he is continuing the LLS grant-funded research in the Starczynowski lab . Starczynowski said Vick has shown exceptional dedication to his work and a passion for advancing the understanding of blood cancers.
“Over the course of his time in the lab, I have seen him grow not just as a researcher but also as a leader,” said Starczynowski, PhD, associate director of Cincinnati Children’s Cancer and Blood Diseases Institute, associate director for basic science research at the University of Cincinnati Cancer Center and a professor in the Department of Pediatrics in UC’s College of Medicine. “He has developed a remarkable ability to approach complex scientific questions with creativity and rigor while also mentoring younger members of our team. His collaborative spirit and willingness to tackle challenges head-on have made him an invaluable asset to our research efforts.”
Vick conducts his research in Daniel Starczynowski's lab at Cincinnati Children's. Photo/Andrew Higley/UC Marketing + Brand.
Vick said he is thankful for the support he has received to help him grow as an early-career investigator.
“I am exceedingly grateful to my mentors Dan Starczynowski, John Byrd and Emily Curran; Cincinnati Children’s; UC; and the members of the Star Lab, without whom none of this would be possible,” he said. “The LLS Career Development Program is an incredible honor — career-defining — and was also validating as a very new physician scientist.”
Starczynowski noted the LLS Career Development Award is a significant achievement for early-career blood cancer researchers, both in providing crucial funding to further their work and as a strong endorsement of their potential.
“For Dr. Vick, this award is a testament to the impact he has already made in leukemia research and the promise he holds as a physician-scientist,” he said. “It highlights his innovative approach to tackling some of the most challenging problems in cancer biology and positions him well for future successes. I have no doubt that Eric will continue to make significant strides in his career, benefiting both patients and the scientific community.”
Impact Lives Here
The University of Cincinnati is leading public urban universities into a new era of innovation and impact. Our faculty, staff and students are saving lives, changing outcomes and bending the future in our city's direction. Next Lives Here.
Featured photo at top of Vick working in the lab. Photo/Andrew Higley/UC Marketing + Brand.
- Faculty Staff
- College of Medicine
- University of Cincinnati Cancer Center
- Academic Health Center
Related Stories
September 6, 2024
The University of Cincinnati Cancer Center’s Eric Vick, MD, PhD, has been awarded a nearly $215,000 grant from the Leukemia and Lymphoma Society (LLS) and a $50,000 American Society of Clinical Oncology (ASCO) Young Investigator Award to continue research into a combination therapy treatment for acute myeloid leukemia (AML).
Ways to cope with the emotional side of breast cancer
The University of Cincinnati's Barbara Walker was featured in an Everyday Health article discussing ways to cope with the emotional aspect of metastatic triple-negative breast cancer.
Chronic skin disorder risk reduces over smoke-free years
September 5, 2024
The University of Cincinnati's Robert Van Haren was featured in a MedCentral article discussing recent research that found smoking cessation significantly reduced the risk of developing skin condition hidradenitis suppurativa.

Mobile phones are not linked to brain cancer, according to a major review of 28 years of research

Director Radiation Research and Advice, Australian Radiation Protection and Nuclear Safety Agency (ARPANSA), and Adjunct Associate Professor, University of Wollongong

Assistant Director, Health Impact Assessment, Australian Radiation Protection and Nuclear Safety Agency (ARPANSA), and Adjunct Associate Professor (Practice), School of Public Health and Preventive Medicine, Monash University
Disclosure statement
Sarah Loughran receives funding from The National Health and Medical Research Council of Australia (NHMRC). She is the Director of Radiation Research and Advice at the Australian Radiation Protection and Nuclear Safety Agency (ARPANSA), a member of the Scientific Expert Group at the International Commission on Non-Ionizing Radiation Protection (ICNIRP), and a member of the World Health Organisation Task Group on Radiofrequency Fields and Health Risks.
Ken Karipidis is the Assistant Director of Health Impact Assessment at the Australian Radiation Protection and Nuclear Safety Agency and he is also the Vice Chair of the International Commission of Non-Ionizing Radiation Protection.
Monash University provides funding as a founding partner of The Conversation AU.
University of Wollongong provides funding as a member of The Conversation AU.
View all partners
A systematic review into the potential health effects from radio wave exposure has shown mobile phones are not linked to brain cancer. The review was commissioned by the World Health Organization and is published today in the journal Environment International .
Mobile phones are often held against the head during use. And they emit radio waves, a type of non-ionising radiation . These two factors are largely why the idea mobile phones might cause brain cancer emerged in the first place.
The possibility that mobile phones might cause cancer has been a long-standing concern. Mobile phones – and wireless tech more broadly – are a major part of our daily lives. So it’s been vital for science to address the safety of radio wave exposure from these devices.
Over the years, the scientific consensus has remained strong – there’s no association between mobile phone radio waves and brain cancer, or health more generally.
Radiation as a possible carcinogen
Despite the consensus, occasional research studies have been published that suggested the possibility of harm.
In 2011, the International Agency for Research on Cancer (IARC) classified radio wave exposure as a possible carcinogen to humans . The meaning of this classification was largely misunderstood and led to some increase in concern.
IARC is part of the World Health Organization. Its classification of radio waves as a possible carcinogen was largely based on limited evidence from human observational studies. Also known as epidemiological studies, they observe the rate of disease and how it may be caused in human populations.
Observational studies are the best tool researchers have to investigate long-term health effects in humans, but the results can often be biased.
The IARC classification relied on previous observational studies where people with brain cancer reported they used a mobile phone more than they actually did. One example of this is known as the INTERPHONE study .
This new systematic review of human observational studies is based on a much larger data set compared to what the IARC examined in 2011.
It includes more recent and more comprehensive studies. This means we can now be more confident that exposure to radio waves from mobile phones or wireless technologies is not associated with an increased risk of brain cancer.

No association
The new review forms part of a series of systematic reviews commissioned by the World Health Organization to look more closely at possible health effects associated with exposure to radio waves.
This systematic review provides the strongest evidence to date that radio waves from wireless technologies are not a hazard to human health.
It is the most comprehensive review on this topic – it considered more than 5,000 studies, of which 63, published between 1994 and 2022, were included in the final analysis. The main reason studies were excluded was that they were not actually relevant; this is very normal with search results from systematic reviews.
No association between mobile phone use and brain cancer, or any other head or neck cancer, was found.
There was also no association with cancer if a person used a mobile phone for ten or more years (prolonged use). How often they used it – either based on the number of calls or the time spent on the phone – also didn’t make a difference.
Importantly, these findings align with previous research . It shows that, although the use of wireless technologies has massively increased in the past few decades, there has been no rise in the incidence of brain cancers.
A good thing
Overall, the results are very reassuring. They mean that our national and international safety limits are protective. Mobile phones emit low-level radio waves below these safety limits, and there is no evidence exposure to these has an impact on human health.
Despite this, it is important that research continues. Technology is developing at a rapid pace. With this development comes the use of radio waves in different ways using different frequencies. It is therefore essential that science continues to ensure radio wave exposure from these technologies remains safe.
The challenge we now face is making sure this new research counteracts the persistent misconceptions and misinformation out there regarding mobile phones and brain cancer.
There remains no evidence of any established health effects from exposures related to mobile phones, and that is a good thing.
- Wireless technology
- Mobile phones
- World Health Organization (WHO)
- Brain cancer
- Mobile technology
- New research, Australia New Zealand
Service Centre Senior Consultant
Director of STEM
Community member - Training Delivery and Development Committee (Volunteer part-time)
Chief Executive Officer
Head of Evidence to Action

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
Current methods in translational cancer research
Michael w. lee.
1 Department of Medical Education, Dell Medical School, University of Texas at Austin, Austin, TX USA
2 Department of Oncology, Dell Medical School, University of Texas at Austin, Austin, TX USA
3 Livestrong Cancer Institutes, Dell Medical School, University of Texas at Austin, Austin, TX USA
Mihailo Miljanic
Todd triplett, craig ramirez, kyaw l. aung, s. gail eckhardt, anna capasso.
Recent developments in pre-clinical screening tools, that more reliably predict the clinical effects and adverse events of candidate therapeutic agents, has ushered in a new era of drug development and screening. However, given the rapid pace with which these models have emerged, the individual merits of these translational research tools warrant careful evaluation in order to furnish clinical researchers with appropriate information to conduct pre-clinical screening in an accelerated and rational manner. This review assesses the predictive utility of both well-established and emerging pre-clinical methods in terms of their suitability as a screening platform for treatment response, ability to represent pharmacodynamic and pharmacokinetic drug properties, and lastly debates the translational limitations and benefits of these models. To this end, we will describe the current literature on cell culture, organoids, in vivo mouse models, and in silico computational approaches. Particular focus will be devoted to discussing gaps and unmet needs in the literature as well as current advancements and innovations achieved in the field, such as co-clinical trials and future avenues for refinement.
Introduction
Extensive efforts directed towards mapping the cancer genome have yielded remarkable insight into the genomic changes that occur during tumorigenesis. Analysis of 2658 whole-cancer genomes from 38 tumor types by the Pan-Cancer Analysis of Whole Genomes (PCAWG), Consortium of the International Cancer Genome Consortium (ICGC), and The Cancer Genome Atlas (TCGA) demonstrated that on average, cancer genomes contain 4–5 driver mutations from coding and non-coding genome elements [ 1 ]. They also found that approximately 5% of tumors had no identifiable driver, suggesting that additional unidentified driver genes exist [ 1 ]. In one of several companion articles published by the ICGC/TCGA/PCAWG, an evolutionary history of the cancers based on the aforementioned sequence data was identified [ 2 ]. Based on these data, it is not far-fetched to conceive that rational deployment of therapeutics or interventions could shift the evolutionary trajectory of the malignant phenotype.
When taken together, the voluminous amount of cancer genome sequencing data that has been generated provides a high-fidelity roadmap of tumorigenesis that can be experimentally exploited to develop a predictive, formulaic method of treating cancer based on mutational changes. But understanding the biological processes of cancer progression is only part of the equation. Knowledge of how the tumor genome changes and adapts upon exposure to therapeutic agents and environmental carcinogens is equally important, as is deciphering the role of epigenetics and protein modifications in oncogenesis. Thus, it will be the role of translational research tools to unravel the complexities of treatment response and resistance, and how this alters the trajectory of tumor development and progression, in the face of genomic changes.
There is an imperative to develop a multi-faceted approach towards modeling cancers in the laboratory that are eminently translatable into the clinical environment. However, experimentally modelling tumor development so that it provides accurate, clinically meaningful, and actionable data to screen patients for risk, treatment selection, and prediction of treatment adverse effects is challenging, in part because tumors are heterogeneous entities.
Tumor heterogeneity and clonal evolution pose formidable barriers to studying cancer biology, immune-tumor interactions, and the response of cancers to therapeutic agents [ 3 – 6 ]. Variations exist between different cancer types in terms of their genetic and epigenetic heterogeneity and, furthermore, clonal evolution can be altered by exposure to chemotherapeutics [ 6 – 8 ]. Tumor heterogeneity is not restricted to the tumor and can extend into the tumor microenvironment, including non-tumor fibroblasts, immune cells, endothelial cells, and matrix components, that can influence propagation of a tumor and its response to therapy [ 9 ].
Therefore, translational models for studying cancer should, ideally, provide an environment for cancers to progress along their natural course of evolution so that tumor heterogeneity can be studied in the presence and absence of therapeutics.
Here, we will explore the current literature covering in vitro tools such as traditional cell line based tissue culture and newer in vitro methods such as 3D organoid models that more accurately simulate the in vivo tumor environment. In vivo modalities such as xenografts and syngeneic mice, genetically engineered mice, and patient-derived xenografts will also be discussed. Lastly, in silico methods will be reviewed with a focus on bioinformatics and computational tools that can be used to model tumor evolution and drug sensitivity. As will be discussed, some of these tools are more aptly suited for exploring tumor evolution and heterogeneity, whereas others are more relevant for studying metastasis, drug discovery, or screening novel compounds.
Cell culture
Cell culture has long been a platform to discover gene alterations in cancer, identify aberrant signaling pathways, and screen new chemical entities as potential chemotherapeutic agents (Fig. (Fig.1). 1 ). While there are many drawbacks and well-known shortcomings of traditional cell culture such as a lack of three dimensional architecture, changes in drug responsiveness, and growth changes with repeated passage, as well as limitations in studying drug metabolism and metastasis; the majority of current knowledge about the biology of cancer cells has been discovered using cell culture. Numerous studies have shown that cell culture systems can model genomic and transcriptomic changes seen in primary tumors [ 10 , 11 ]. For example, Barrentina et al. used DNA copy number and gene expression patterns to determine equivalency between a human cell line library, with 947 cell lines, and primary tumors from corresponding tissues [ 11 ]. They reported positive correlations for DNA copy number ( r = 0.77), gene expression patterns ( r = 0.60), and point mutation frequency ( r = 0.71) for all but a small number of the cell lines examined [ 11 ]. On the basis of the gene expression profiles, subsequent pharmacological interrogation of this cell line library revealed several previously unrecognized genes and cell features that correlate with drug response. For example, expression of the aryl hydrocarbon receptor (AHR) was found to correlate with enhanced response to MEK inhibitors in NRAS mutant cell lines [ 11 ].

Comparison of strengths and weaknesses of current models for translational cancer research . For each model, the number of clocks and dollar signs correspond to the preparation time and relative cost of establishing and maintaining the model. Likewise, color coding indicates the degree to which the model is suited for a particular type of translational research (dark red denoting poorly suited to dark green denoting well suited)
However, a central issue with cell culture systems is that, although driver mutations are generally preserved, prolonged culturing can lead to secondary genomic changes including copy number variations and transcriptomic drifts [ 12 , 13 ]. Indeed, these types of culture-condition-induced alterations can lead to changes in multi-drug resistance genes that differ from clinical samples. This observation was first made while evaluating over 80 samples of untreated primary ovarian carcinoma in comparison with additional cancer types from the NCI-60 panel and evaluating the expression profile of over 380 MDR-related genes [ 7 ]. These authors expanded their analysis to include several other cancer types including colorectal cancer, breast cancer, metastatic melanoma, and glioblastoma, which similarly demonstrated that cultured cell pairs from a primary tumor bore more resemblance to each other than pairs from different primary tumors of the same origin [ 7 ]. This indicates that cultured cells can retain genomic signatures from the primary tumor despite the influence of in vitro culture conditions.
In addition to the effects of prolonged culture conditions, there are also other drawbacks of traditional 2D cell systems. For example, the absence of extracellular architecture, including stromal cells and matrix components, can alter innate biological processes of cultured cancer cells and modify their response to therapeutics [ 14 ]. It has been shown that inclusion of tumor stromal cells and extracellular matrix mediators can drive tumor growth, stimulate angiogenesis, favor an inflammatory environment, and promote drug resistance [ 10 , 14 – 16 ]. For these and other reasons, 3D tissue culture and organoid systems were developed.
There are a number of studies that have demonstrated the impact that incorporating stromal cells and matrix components can have on the response of cancer cells to therapeutics [ 17 – 19 ]. In some cases, this involved developing novel culture systems. For example, a recently published study described the development of a 3D tumor invasion model that utilizes traditional cell culture together with a customized system that incorporates extracellular matrix (ECM) [ 20 ]. In this study, the authors used highly metastatic pancreatic ductal adenocarcinoma (PDAC) cells as their model. Briefly, a fabricated platform was developed with posts coated in ECM and arranged in a 96-well format upon which tumor cells suspended in type I collagen oligomer were seeded. Following initial polymerization of the oligomer, a plug-like tumor compartment at the bottom of the 96-well plate formed onto which media, drugs, and cancer-associated fibroblasts (CAFs) could be overlaid. They noted that the addition of the CAFs resulted in dramatic changes in the phenotype of both PDAC cells and CAFs together with matrix remodeling and, importantly, pronounced invasion into the surrounding matrix [ 20 ]. Markers of epithelial to mesenchymal transition (EMT) were also examined and suggested an EMT-independent invasion phenotype [ 20 ]. A proof-of-concept drug screening treatment regimen with 10 different doses of gemcitabine was performed using Hoechst 33342, Click-iT EdU, and Mito Tracker Red (to assess nuclear changes/condensation, proliferation, and mitochondrial metabolism, respectively). Their results revealed effects of gemcitabine on cell proliferation, although with only moderate effects on invasion. Based on these data, the authors were able to establish initial validation of this system as a potentially viable drug screening platform. They also noted that this system has distinct advantages over other established models of migration/invasion/metastasis such as scratch assays, transwell (Boyden chambers), and 3D spheroid invasion assays in terms of standardization of spheroid and matrix components for high throughput/high content screening [ 20 ].
Another drawback of using cell culture as a clinically predictive model is the minimal degree of drug metabolism that occurs in a single-cell lineage context. Drug metabolism, particularly cytochrome P450–mediated drug metabolism, may yield a mixture of active and inactive metabolites that ultimately contributes to the pharmacological efficacy of chemotherapeutic agents under investigation [ 21 ]. This complexity of metabolism is lost in cell culture systems. Furthermore, the lack of a normal control for tissue culture is an important factor [ 22 ].
Although isogenic cell lines can be generated using homologous recombination to provide a comparator cell line when analyzing single gene differences, this technique is still hampered by cell culture effects [ 21 ]. Studies have demonstrated that there can be notable differences in cell expansion and drug sensitivity between identical isogenic cell lines as a result of 2D or 3D conditions [ 23 ]. For example, the isogenic DLD1 KRAS +/− , KRAS G13D/− , PIK3CA +/− , and PIK3CA E545K/− colorectal cancer cell lines exhibited substantially different growth kinetics and sensitivity to the MEK inhibitor PD 0325901 depending on whether cells were cultured in 2D or 3D conditions despite their identical, isogenic status [ 23 ]. Due to these limitations, other methods of tissue culture have emerged that more closely recapitulate clinical heterogeneity while limiting artificial cell culture effects [ 22 ].
Organoids are an advancement of traditional tissue culture that is meant to more closely mimic the 3D architecture of primary tumors. Hans Clevers defined an organoid as “a 3D structure grown from stem cells and consisting of organ-specific cell types that self-organize through cell sorting and spatially restricted lineage commitment” [ 24 ]. As noted in this conventional definition, organoids can be derived from embryonic, adult, or pluripotent stem cells [ 25 ]. However, somatic cells can also be conditionally reprogrammed and cultured as organoids using feeder cells and Ras homolog (RHO) kinase inhibitors [ 25 ]. This method is not to be confused with somatic cell reprogramming which involves generation of induced pluripotent stem cell (iPSCs) from somatic cells using techniques such as somatic cell nuclear transfer (SCNT), cell-cell fusion, exposure to extracts of pluripotent cells, or iPSC technology [ 26 ]. It has been previously shown that organoids may be established from a variety of tumor types, such as colon, pancreas, esophageal, liver, endometrial, breast, and prostate cancers all requiring different composition of cultural media [ 27 ]. Previous studies have shown that organoids can maintain similar histopathological features derived from the primary tumor not only in the in vitro setting but also after being injected into immunocompromised mice permitting their use as an efficient tool to validate drug responses obtained in vitro and also in more complex in vivo systems [ 27 ]. In fact, their ability to better recapitulate tumor structure may have a greater impact on predicting responses to novel and conventional anti-cancer therapeutics with respect to 2D cell lines, opening an avenue towards drug development and personalized medicine [ 27 ]. Organoids are not without downsides, however. As will be discussed, they are slightly more technically and time intensive than traditional cell culture, can be subject to overgrowth and passage effects, and have limitations similar to cell culture relating to drug metabolism.
In general, organoids have a number of important features that set them apart from traditional cell culture and animal models [ 22 , 28 ]. They self-organize and mimic the general architecture of the tissue of origin, and, importantly, maintain these characteristics over successive passages. This more relevant in vitro model offers advantages for studying tumor progression, treatment responsiveness, and interactions with the immune system and the tumor microenvironment (Fig. (Fig.1). 1 ). Moreover, the morphological stability of organoids allows them to be coupled with other powerful techniques such as CRISPR/Cas9 and single-cell analysis [ 28 ]. As will be discussed below, organoids are also genetically stable models [ 24 , 29 ].
The most common method of generating organoids from normal and tumor tissue is with adult stems cells isolated from resected tissue or biopsies using conditioned media supplemented with growth and selection factors [ 24 , 25 ]. Most organoid media is supplemented with R-spondin, Wnt, epidermal growth factor (EGF), and Noggin, together with the ALK (anaplastic lymphoma kinase) inhibitor A83-01, p38 inhibitor SB202190, and nicotinamide [ 28 ]. Lgr5 is a G protein–coupled receptor that is found on stem cells and binds R-spondin, whereas Wnt (i.e., Wnt-3A) is a ligand for Frizzled receptors found on Lgr5 + stem cells [ 28 ]. Noggin is included because it is a bone morphogenic protein (BMP) receptor inhibitor. BMP receptor engagement on Lgr5 + stem cells negatively regulates stemness, whereas EGF binding to EGF receptors on Lgr5 + stem cells increase stemness [ 28 ]. A83-01 and SB202190 both appear to increase the number of passages of organoids and their long-term culture [ 29 ]. This method of culturing adult stem cells has been validated in a variety of tissue and tumor types although important differences exist between tissues in terms of specific composition of the growth media [ 24 ]. In addition to distinct culture requirements, organoid culture success rates can vary significantly between different cancer types [ 30 ].
As noted, adult stem cell–derived organoids are more frequently employed and appear to have a number of advantages over pluripotent stem cell organoids in terms of retention of phenotypic tissue features, biobanking, genetic modification, generation of matched normal controls, and incorporation of an immune system, among others [ 22 ].
However, a central question to consider with organoids, given the dynamic and heterogeneous nature of the cancer genome, is how well do organoids reflect the genetic and mutational profile of the parent tumor and how stable are the genetics over the study/treatment period? In other words, do the conditioned and semi-artificial culture conditions of organoid growth environments result in deviation of tumors from their inherent genetic mutational evolution?
Modeling tumorigenesis and tumor evolution
As noted, intensified focus on the evolution and development of tumors has created a need to craft systems capable of modeling these processes [ 28 ]. Organoids appear to be a flexible system in terms of genetic manipulation and therefore can be used as a platform for discovery of novel genes involved in tumorigenesis [ 31 ]. Building on a previous study that used a Sleeping Beauty transposon–based mutagenesis screening system, Takeda et al. used a CRISPR-Cas9 gene-editing strategy to knockout genes in intestinal organoids derived from both mice and from human colorectal tumors [ 32 , 33 ]. This system has significant advantages over knockout mouse models in terms of assessing cancer driver gene function, particularly in terms of cost and time. From a pool of genes they identified, they selected 29 candidate tumor suppressor genes (including Trp53 , Smad4 , and Pten ) for loss-of-function studies. Using organoids sourced from mouse intestinal tumors with a mutated APC and mutated Kras background (APC Δ716 , Kras +/G12D ) (both commonly mutated genes in colorectal cancer), they transduced the organoids with lentiviral vectors containing Cas9 and GFP, followed by lentivirus transduction with viral particles containing pools of tumor suppressor candidate gene gRNAs. Once these pooled, loss-of-function, organoid models were established, they were transplanted into NOD/SCID/γ-chain (NSG) mice for subsequent studies. When taken together, they revealed that loss of function of one or more genes leads to liver metastasis in mice orthotopically implanted with these organoids [ 33 ]. Importantly, the system they developed for colorectal cancer is an innovative strategy for manipulating driver genes using organoids that can be leveraged for discovery of other critical driver genes and assessment of therapeutics.
Organoids have also been established from multiple single cells isolated from both normal intestinal crypts and colorectal cancers [ 34 ]. In this study, pieces of tumors were isolated from distinct locations of colorectal tumors from three previously untreated patients, grown as organoids, and sorted via flow cytometry to yield single cells for subsequent organoid culture. Phylogenetic trees were constructed using mutational data derived from whole genome sequencing or targeted gene panel sequencing, comprising 360 known cancer genes. Methylation patterns, epigenetic analysis, and drug sensitivity were also examined for the single cell–derived organoids. Interestingly, single cell–derived organoids exhibited extensive genetic heterogeneity. Key putative driver mutations identified as being in the trunk of the phylogenetic tree of one patient included PIK3A (E81K) and BRAF (V600E) together with microsatellite instability and hypermethylation of the MLH1 gene. The second patient had two protein truncating mutations in APC together with a mutated TP53 containing a homozygous splice site mutation [ 34 ]. The third patient, on the other hand, had a mutated KRAS (A146T) and two truncating APC mutations. Based on the mutational load and the somatic mutations observed, mutational signatures were identified and applied to each segment of the phylogenetic tree for each of the single cell–derived organoids [ 34 ]. Thus, these findings provide additional support for the concept that as cancers develop and evolve, they acquire more somatic mutations compared to normal cells. The processes that permit successive mutation in these colorectal cancer cells likely become more permissive over time; however, the timing remains unclear. Interestingly, as noted above, two of the patient-derived organoids were mismatch-repair proficient [ 34 ].
The aforementioned study demonstrates that organoid based systems can be used to construct complex phylogenetic and mutational signature profiles of single cells that appear to recapitulate the mutational dynamics of the tumor. Thus, organoids may be used map mutational processes that could predict tumor responsiveness to the environment or therapeutics over time.
Platform for drug screening and drug discovery
In addition to their utility in dissecting tumorigenesis and cancer evolution, organoid models can be used to study a tumors response to cancer chemotherapeutic agents.
This generally entails constructing a library or biobank of organoids established from numerous tumor samples or biopsies and performing gene expression profiling prior to drug screening, which is technically complex. The goal is to establish a protocol for creating a patient-derived organoid (PDO) tumor model that faithfully reproduces the genotype, phenotype, and therapeutic response of the patient’s tumor such that it can be used to study new and existing agents, as well as drug resistance.
The combination of gene expression analysis and therapeutic profiling is now more readily being employed to characterize and validate organoids in order to match their biological progression response to treatment with that of the tumor [ 35 ]. Studies differ in several important terms that need to be considered: the size of the biobank or library, the outgrowth efficiency from the primary tumor sample, culture conditions, methods used to characterize organoid gene expression, similarity to the primary tumor, the number and types of agents screened, and the therapeutic outcomes.
In general, the size of biobanks or libraries is limited by the availability of tumor samples. Additionally, the method of tumor tissue procurement (i.e., biopsy, surgical resection) can alter the size of the biobank or library due to differences in efficiency of organoid outgrowth and isolation techniques [ 36 ]. For example, a recent study constructed a biobank from 83 tumor samples isolated via a combination of surgical resection and biopsy [ 37 ]. The overall outgrowth efficiency of the organoids was 62% [ 38 ]. The majority of the organoids sequenced were obtained via surgical resection, whereas the outgrowth efficiency of biopsy sourced organoids was low (31%) [ 37 ].
Studies on refining the method of establishing organoids from biopsy samples have led to improved outgrowth efficiency [ 36 ]. Starting with 159 pancreatic tumor samples from 138 patients, Tiriac et al. ultimately constructed a library of 114 PDOs from 101 of these patients [ 28 ]. Slight variations in efficiency depending on the route by which the tumor sample was obtained (fine needle biopsy or tumor resection, 72% versus 78%, respectively) were observed. In addition to the patient-derived tumor samples, the authors developed 11 human normal pancreatic ductal organoids from pancreatic islet transplant samples [ 35 ].
Biobanks of organoids can also be used to perform co-clinical trials (a side by side study matching patient drug responses to the drug responses of pre-clinical, translational laboratory models) [ 37 ]. In this study, metastatic colorectal cancer ( n = 16), gastroesophageal cancer ( n = 4), and cholangiocarcinoma ( n = 1) were procured via biopsy and grown as organoids, with a 70% efficiency [ 32 ]. This biobank then served as a platform for drug screening, discussed more below. In a similar study, Ooft et al. examined the predictive potential of organoids derived from metastatic colorectal cancer biopsies ( n = 67) from patients ( n = 61) prior to receiving chemotherapy and compared the patient’s response to drug with that of the organoid’s response to drug [ 39 ]. Due to a variety of circumstances (i.e., retrieval of tissue and cells, quality control, and bacterial infection), the authors report that they achieved a 63% PDO culture rate for tumors isolated from patients via biopsy.
In concert with the route of procurement, culture conditions can also have a profound influence on outgrowth efficiency. Serum-free culture conditions can be used for organoid propagation to ensure that nonepithelial cells do not survive culture and propagation efficiency is maximized [ 35 , 40 , 41 ]. Others have looked at how different types of organoid media can be formulated to select for organoids harboring certain oncogenic mutations while eliminating overgrowth of non-tumor cells [ 37 ]. For example, conditioned organoid media created selective pressure for outgrowth of KRAS G12R mutant tumor cells from normal pancreatic tissue from an unidentified pre-cancerous lesion in a patient sample [ 37 ]. Others have reported growing their PDOs on Matrigel using standard PDO culture media to select LGR5 + stem cells from their biopsies with a high rate of efficiency (70%) in line with previously published data [ 42 ]. Histological and immunohistochemical assessment is often used to confirm that the organoids retain parental tumor characteristics [ 32 ].
Studies are increasingly using combinations of high-throughput methods to document the landscape of gene expression and genetic mutations in organoids as a prelude to interrogating them with drugs. For example, single nucleotide variants (SNVs) and copy number alterations (CNAs), characterized using Sanger sequencing and whole exome sequencing (WES), have been used to establish the genetic and mutational landscape of the organoids PDAC organoids [ 28 ]. In this study, whole genome sequencing (WGS) was employed with a subset of the PDAC-confirmed PDOs to determine the degree of similarity between the organoids and matched tumors [ 28 ]. Similarly, transcriptomic profiling using RNA sequencing (RNA-seq) has been used to compare the gene expression of the PDOs with classic and basal signatures identified from virtually microdissected PDAC [ 35 , 43 ]. WGS and next-generation sequencing (NGS) are also used to look at panels of oncogene or tumor suppressors commonly found in a tumor type to validate the organoid model and to guide selection of therapeutics for experimentation [ 32 , 37 ].
Ultimately, the end goal of these efforts to characterize organoids is to understand the parameters governing sensitivity or resistance to conventionally used drugs and to discover new agents. Indeed, transcriptomic gene expression profiling is facilitating screening of difficult to treat cancers such as PDAC [ 28 ]. Pharmacotyping PDAC PDOs using commonly employed conventional chemotherapeutic agents such as gemcitabine, nab-paclitaxel, irinotecan, 5-fluoruracil, and oxaliplatin revealed interpatient variability to these agents [ 28 ]. This, in turn, allowed further classification of the PDOs according to degree of chemosensitivity (i.e., most responsive, intermediately responsive, and least responsive). Comparison of these results with retrospective treatment data for patients, from which these PDOs were derived, revealed that the treatment responses as assessed by progression-free survival were similar [ 35 ]. Subsequently, the effectiveness of a range of targeted agents was assessed, with several demonstrating efficacy towards chemoresistant PDOs, including selumetinib (MEK 1/2 inhibitor), afatinib (EGFR tyrosine kinase inhibitor), everolimus (mTOR inhibitor), and LY2874455 (FGFR inhibitor) [ 35 ]. Thus, incorporating transcriptomic data with drug sensitivity pharmacotyping data provides further stratification of the PDOs and may enable development of novel therapeutic strategies.
Biobanks of organoids can also be used to screen large sets of conventional and targeted agents in tandem with gene expression profiling. Following gene expression profiling of hundreds of genes known to be involved in PDAC oncogenesis, the viability of a bank of PDAC derived organoids was assessed following treatment with a range of agents [ 37 ]. In this case, twenty-four PDOs in this bank were used to screen a panel of 76 therapeutic agents revealing a wide range of individual responses to targeted therapeutics and, in the case of a small subset of 4 patients, the PDO responses correlated with clinical responses [ 37 ]. Importantly, they queried their PDO system with a novel therapeutic, protein arginine methyltransferase 5 (PRMT5) inhibitor EZP01556 that exploits a synthetic lethal vulnerability of the 80–90% of PDACs that are deficient for the gene MTAP (methyladenosine phosphorylase) [ 44 ]. Indeed, MTAP organoid lines exhibited greater sensitivity to PRMT5 inhibitors, although some subsets of MTAP + organoids also responded [ 37 ]. This both reinforces the potential of organoid systems for testing novel agents and underscores the need for further study.
Finally, screens of drugs can be done in organoids using treatment protocols that mimic phased clinical trials or use a co-clinical trial design to explore combination therapy [ 32 , 35 ]. Using a library of 55 drugs in phase I–III clinical trials or currently in clinical use, Vlachogiannis et al. demonstrated the ability of their PDO system to recapitulate drug responses by correlating drug responses of the PDO to that of the patient and tumor genotype [ 37 ]. While they found that the PDOs with amplifications in some genes responded to agents targeting the products of those genes, they did note that not all mutations profiled were predictive of response. For example, PI3K mutations did not predict response to the dual PI3K/mTOR inhibitor GDC-0980 [ 37 ]. As part of the co-clinical trial, they established orthotopic xenograft mice using organoids and assessed the response to regorafenib, a multi-angiogenic kinase inhibitor [ 37 , 45 ]. Using MRI imaging in tandem with CD31 immunostaining, they observed a similar response in patients (resistance vs prolonged disease stability) compared with PDOs derived from these patients. They report that their PDO system exhibited 100% sensitivity, 93% specificity, a positive predictive value of 88%, and negative predictive value of 100% to targeted agents or chemotherapy, suggesting the potential utility of such pre-clinical systems for drug screening and activity prediction [ 37 ].
In another recent example, CRC PDOs were randomized into treatment arms: 16 PDOs to standard first-line therapy with 5-FU and oxaliplatin; 12 PDOs to second-line therapy with 5-FU and irinotecan; 10 PDOs to single-agent irinotecan. Following this, they created a classification model to predict non-responders to monotherapy with irinotecan. Analysis of growth rate inhibition metrics and dose response curves for PDOs (representing both progressive disease and stable disease) after treatment with irinotecan provided them with a training data set they could query using a model prediction method called leave-one-out-cross validation (LOOCV) [ 39 ]. This allowed them to classify 80% of non-responders from organoid drug sensitivity data compared with the corresponding patient source. They note that this assay only required 5000 cells to perform. Similar classification of combination therapy with 5-FU and irinotecan also suggested that PDOs have predictive value for combination therapy. In this case, to generate the complementary dose response curves for the 5-FU and irinotecan analysis only required 10,000 cells and could be generated in 21 days, a vast improvement over traditional cell culture systems [ 39 ]. Interestingly, they showed that their organoid system failed to predict response to the combination of 5-FU and oxaliplatin. This is an important result because, as they note, it reveals the limitations in modeling combination therapy responses in organoids [ 35 ]. This may be a result of incomplete understanding into the nature of the synergism between agents or could also be due to a lack of certain biological components in the organoid system such as metabolizing enzymes (i.e., cytochrome P50s) or the microbiome.
As discussed, many studies have focused on validating the organoid models, building biobanks, or libraries, and screening them with a small cadre of predominately known therapeutic agents [ 28 ]. However, what remains to be determined is whether organoid-based systems can actually facilitate the discovery of novel (and active) therapeutic agents. Creation of novel systems or strategies to propagate and curate organoids for drug screening may provide an avenue to successful identification of novel cancer drugs. Indeed, a novel miniaturized organoid culture method that incorporates mini-rings for 3D culture that can be rapidly screened for drug sensitivity (within 1 week from surgical resection) was recently described [ 46 ]. This system uses fewer cells, smaller amounts of Matrigel, and entails seeding cells around the rim of a 96-well plate in a ring shape using a single well or multi-channel pipette. Additional advantages of this system are that adding and removing media and drugs can be done with minimal disruption to the cells and the organoids appear to resemble the tumor from which they were derived. Small-scale proof-of-concept experiments using doxorubicin, staurosporine, and a novel peptide inhibitor of p53, ReACp53, revealed that the Matrigel layer allowed both small molecules and peptides to penetrate and reach the organoids. A larger screen with 240 protein kinases, at two different concentrations, was performed using organoids derived from 4 patients with ovarian and peritoneal tumors. These included inhibitors of CDK, MEK, EGFR, PI3K/mTOR, IKK, HDAC, and Flt [ 46 ]. In general, they reported tumor-specific, non-redundant responses for the inhibitors they assayed with the exception of BGT226 (an PI3K/mTOR inhibitor) which elicited responses from all of the organoids screened. Thus, in accordance with the other studies discussed so far, organoids appear to be a reproducible platform for personalized drug screening that appropriately recapitulates the challenges of inter-and intra-tumor heterogeneity observed in patients. This system, which does appear cheaper and faster, could potentially be deployed at a scale which could yield more statistically robust and clinically translatable results.
The recently reported use of single-cell techniques to establish organoid cultures is another promising approach to determine if the intra-tumor heterogeneity seen in individual cancer cells translates into differential treatment responsiveness [ 34 ]. This is supported by the observation that organoids can stably retain the genetic and epigenetic during propagation [ 29 ]. As a consequence, derivation of organoids from single-cells can shed light on how drug resistance develops in tumors, which may occur late in tumorigenesis at geographically distinct sites in the tumor [ 27 ].
The advent of organoids-on-a-chip, which seek to replicate in vivo conditions in vitro , using organoids is particularly exciting [ 47 ]. This technology offers the opportunity to assess cancer cell interaction with normal tissue, the immune system, and response to therapeutics. A model to study the interactions of T cells with tumor organoids established from mismatch-repair deficient CRC or non-small-cell lung cancer (NSCLC) was recently reported [ 48 ]. In this system, co-culture of peripheral blood lymphocytes with tumor organoids led to enhanced T cell tumor reactivity and cell killing [ 48 ]. Furthermore, T cell reactivity was exclusive to the organoids that had been previously co-cultured, indicating and that this system may be utilized to assess the extent of immune-specific cytolytic T cell–mediated tumor destruction [ 48 ]. Other studies also lend support to the use of organoids in elucidating immune-tumor interactions further suggesting that organoid models could be adapted to an organoid-on-a-chip format together with stromal cells and microvasculature [ 42 ].
Another application of organoids-on-a-chip technology could be in assessing drug-induced target and non-target organ toxicity, a frequent cause for clinical drug failure [ 47 , 49 ]. For example, Skardal et al. created a triple-tissue organ-on-a-chip platform in which they integrated heart, liver, and lung organoid systems through perfusion-driven microreactors constructed on poly-dimethyl siloxane that was connected serially under fluid flow [ 47 , 49 ]. In the “liver-on-a-chip” component of this integrated system, they show stable and reproducible activity of stellate cells, Kupffer cells, and hepatocytes with measurable ATP production. Remarkably, this system can also be used to measure output of products such as albumin and urea, harbors metabolically active cytochrome P450 enzymes (CYP3A4 and CYP2C19), and it can be used to assess liver damage following exposure to acetaminophen with lactate dehydrogenase levels and liver cell protein release as outputs of drug-induced related liver toxicity [ 47 , 49 ]. This last point is crucial, as this team demonstrated superior advantages of this system compared with 2D hepatocyte culture, and highlights the clinical relevance of this novel approach in predicting liver metabolism and toxicity which are critical components of the drug development process [ 47 , 49 ].
Finally, an area of need that could be addressed with organoids-on-a-chip is modeling the effect that biochemical and metabolic intermediates have on tumors and their extracellular environment. In other words, can the non-cellular conditions in which a tumor resides and proliferates be recapitulated? Some promising work along these lines has appeared in the literature in regard to tumor microenvironment metabolites but more progress is needed [ 50 , 51 ]. Advancements in microfluidics and perfusable blood vessels permit incorporation of soluble factors and nutrients, normally present in the extracellular milieu, into these models that more accurately mimic in vivo culture conditions and tumor microenvironments [ 47 , 52 ].
In summary, organoids, like the human patients they are derived from, exhibit genotypic and phenotypic heterogeneity, in some cases, organoids recapitulate clinical responses. Establishment of organoid biobanks and libraries allows high-throughput compound screening that can be readily translated into animal models.
Mouse models
Tumor development is, in general, a progressive process fueled by mutations in driver genes such as oncogenes or tumor suppressor genes that ultimately provide an evolutionary advantage for tumor survival [ 53 , 54 ]. Considering the number of driver genes that can be mutated, it is not surprising that tumors are heterogeneous, containing many sub-clones that take different paths during clonal evolution in a self-selecting, self-propagating manner. In addition to genetic changes, the tumor microenvironment, the immune system, exogenous toxins and environmental xenobiotics, cancer chemotherapy, and the microbiota can all influence the mutational course a tumor traverses as it grows and evolves. Therefore, having a model system whereby biological consequences of step-wise mutational changes can be mapped, and future mutations predicted, is of great importance. Gaining insight into this process could aid in drug discovery and tailored treatment of patients. In the sections that follow, we will review the different mouse models currently used in cancer research and discuss representative studies, advantages, and disadvantages of each system. For each of the models, we will consider its suitability for studying: tumor evolution and heterogeneity, metastasis, immune-tumor interactions, and suitability as a platform for drug discovery and screening (Fig. (Fig.1 1 ).
Xenograft and syngeneic mouse models
For decades, the most basic and frequently employed mouse models used to assess tumor growth and screen conventional chemotherapy or candidate drugs have been simple xenografted or syngeneic mice. Typically, these mouse models involve subcutaneous administration of human (xenograft) and mouse (syngeneic) tumor cells without regard to the organ of tumor origin (heterotopic) or via implantation of tumor tissue or cells into the tissue corresponding to the site of the tumors origin (orthotopic) [ 55 ]. Cell culture–derived xenograft mice and syngeneic mice require less technical skill and time to establish but they are less predictive of a patient’s response to therapeutics compared with genetically engineered mouse models (GEMMs) or patient-derived xenograft (PDX) mice, which are discussed in subsequent sections [ 56 ].
Tumor evolution and heterogeneity
For a variety of reasons, simple subcutaneous xenograft mouse models, where human tumor cells grown on a plate are injected into a mouse, are not robust tools in investigating tumor evolution [ 55 , 57 ]. This is, in part, due to the source material. As discussed, cancer cells grown on plates are unreliable predictors of mutational progression in tumors due to numerous additional mutations cell lines acquire from repeated propagation on artificial tissue culture plates [ 12 , 58 ]. Indeed, cancer cell lines tend to lose their heterogeneity and become more homogeneous with continued selection on cell culture media in the absence of tumor microenvironment and immune influences [ 57 , 59 ]. Orthotopically implanting human or mouse cancer cells into a mouse yields a more representative model of tumor development because the implanted tumor is placed in the organ environment similar to that from which it originally arose [ 55 , 57 ]. In the case of some tumor types, such as breast cancer, the orthotopically implanted tumor fragments are more representative models of tumor development and tumor microenvironment [ 55 , 57 ].
Syngeneic mouse models represent an improvement on heterotopic subcutaneous or orthotopic xenograft models using human cancer cells to study tumor evolution and tumor heterogeneity because murine tumor cells can be inoculated or implanted into immunocompetent mice [ 60 ]. Syngeneic mice were among the first in vivo oncologic models created, with the discovery of the mouse leukemia model L1210 in 1960 followed closely by establishment of solid tumor syngeneic models [ 58 ]. This leukemia model, studied extensively by Howard Skipper for drug and pharmacokinetic screening, demonstrated not only that combination chemotherapy could be superior to single treatment experiments, but also that the schedule of administration was important to the efficacy of a given regimen. In 1964, Skipper would report the cure of murine leukemia in L1210, a discovery that aided in the development of a durable cure for acute lymphocytic leukemia in a subset of human patients [ 61 ]. Although many years have passed since this milestone, syngeneic mice continue to be used as a pre-clinical models for drug screening, and more recently as essential tools for predicting response to immunotherapy [ 58 , 61 – 63 ]. With their wide application potential, it is important to consider specific molecular features when selecting a model for drug screening, pharmacokinetic studies, and translational potential.
Unfortunately, syngeneic mouse tumors have the disadvantage of a lower mutational load than comparable human tumors [ 57 ]. In general, mouse tumors are less heterogeneous than human tumors because of inter-species differences [ 9 , 64 ]. This ultimately translates into differences in gene expression profiles, baseline immune infiltrates, and response to drug treatment including immune checkpoint blockade [ 58 , 61 ]. Indeed, analyses of syngeneic tumors of colon, breast, renal, and melanoma origin have revealed that there are profound differences between models even of the same cancer type, indicating that extensive profiling of syngeneic models may be necessary for creation of more predictive pre-clinical models [ 61 ]. In a study of 12 different syngeneic orthotopic models of metastatic breast cancer, differences in gene expression profiles, histopathology, angiogenesis, and proliferation rates have also been noted [ 65 ]. Despite this inter-model heterogeneity, these syngeneic mice recapitulated human samples and more than half of the most commonly mutated genes in human breast cancer were represented within their panel and could assist in predictive modeling for different forms of breast cancer [ 65 ].
The suitability of mouse models for studying metastasis continues to be a challenging area. Unsurprisingly, heterotopic mouse models where cancer cell lines are injected into a mouse rarely result in metastasis [ 65 – 67 ]. This is, in part, a product of poor mismatch between the cancer cells and the tumor microenvironment of the mouse and the lack of an immune system (i.e., nude mice and NSG mice) [ 55 , 66 , 67 ]. This is especially true for xenograft models, where human cancer cells are injected into a mouse; however, similarly poor rates of spontaneous metastasis are encountered with allograft injection of mouse cancer cells into mice [ 66 , 67 ]. Interestingly, in the case of intravenous injection, the site of injection dictates to some degree the extent and site of metastasis [ 66 ]. Injection of cancers cells in the tail vein of the mouse results in metastasis to the lung, portal vein injection leads to hepatic metastasis, intracardiac injection yields more diffuse metastasis to brain and bone, and so on [ 66 , 67 ]. Thus, it stands to be questioned if this truly represents metastasis, as the early steps in metastatic development and progression are bypassed due to the route of administration [ 66 , 67 ]. There are, however, a handful of examples where injection of human cancer cells leads to metastasis. These include metastatic prone human cancer cells lines such as MCF-7 and MDA-MB-231 breast cancer cells, KM12 colon cancer cells, A7 and B16 melanoma cells, PC-3 prostate cancer cells, and SKOV3 ovarian cancer cells [ 66 ].
In contrast to heterotopic administration of cancer cells, orthotopically implanting human cancer cells or mouse tumors cells into mice more closely approximates the tumor microenvironment and can, in certain syngeneic mouse models, result in metastasis [ 65 ]. There are a number of mouse cell lines that lead to tumor formation and metastasis such as 4T1, B16, Lewis lung carcinoma cells, Met-1, and RM1 [ 66 ]. It was recently reported using orthotopic injection of 4T1 mouse mammary tumor cells (generated from CARMIL1-WT or CARMIL1-AA cells) into BALB/c mice to study the role of macropinocytosis in mediating treatment resistance [ 68 ]. They found that macropinocytosis fuels tumor growth (possibly by generating intracellular cell nutrients such as amino acids, sugars, fatty acids, and nucleotides via necrocytosis) and increases resistance to 5-FU [ 68 ]. Despite these examples, it is still not clear whether these models are more predictive when utilized to assess anti-cancer therapy.
Immune-tumor interactions
Of all of the models discussed, xenografts are the least useful for studying immune-tumor interactions [ 66 ]. Syngeneic mouse models do have an intact immune system, although it is a mouse immune system and may not approximate the types of interactions observed in human tumor environments (particularly in regards to evaluating biologics targeting human proteins such as PD1) [ 69 ]. Nevertheless, there are advantages to using parenterally or orthotopically derived syngeneic mice including short latency periods, reproducibility, and genetic tractability [ 55 , 70 ]. There are many examples of syngeneic mouse models that have been used to explore immune-tumor interactions [ 71 – 74 ]. To address potential differences between these models for immunotherapy, Mosley et al. meticulously compared syngeneic mouse models through the lens of immunologically hot and immunologically cold tumors that involved gene expression profiling of immune-related pathways and responses to immune checkpoint inhibitors [ 71 ]. The hot or cold phenotype correlates with the extent of T cell infiltration into to the tumor microenvironment [ 75 , 76 ]. They began by assessing the effectiveness of inhibiting immune checkpoints with anti-CTLA-4 antibodies or anti-PD-L1 antibodies in six different syngeneic mouse tumor models (CT26, RENCA, 4T1, B16F10 AP-3, LL/2, and MC38) and observed that only two models responded to the anti-CTLA-4 treatment, CT26 (a mouse colon carcinoma model), and RENCA (a mouse renal cell carcinoma model), as measured by decreases in tumor volume [ 64 ]. Only the CT26 mice responded to anti-PD-L1 treatment. Based on this differential response, the authors performed genomic analysis of the original cell lines used for these mouse models by examining CNVs, whole-exosome analysis, and transcriptomics to look for differences in gene expression signatures and mutational profile between the different models. They discovered that CNV levels in the parent cell lines were not altered by their method of generation and that, in the case of CT26, there was no difference in the overall mutational burden [ 71 ]. Using a panel of 64 prominent cancer genes, the authors then used WES to compare the mutational profile of tumor cell lines with matched tumors. In the case of CT26, they found that APC and Kras were mutated in both CT26 cell lines and human colorectal tumors but that CT26 did not have the Trp53 mutation found in human colorectal tumors. Nevertheless, they found a high overall correlation ( r = 0.766) in mutant allele frequency between the murine cell lines and the corresponding syngeneic murine tumors ( in vitro versus in vivo ) [ 71 ]. Transcriptomic profiling revealed that differences in gene expression in innate immune-related pathways could have accounted for the lack of responsiveness to checkpoint blockade and the cold phenotype of B16 mouse tumors. Further support for differences in the immune system microenvironment between these models came from analysis of immune cell infiltration in tumors via flow cytometry where they profiled nine nonoverlapping innate and adaptive immune cell phenotypes [ 71 ]. This analysis demonstrated that immune cell infiltration varied markedly between the models perhaps accounting for the differential responses. For example, 4T1, MC38, and LL/2 were enriched for immunosuppressive granulocyte macrophage–derived suppressor cells (gMDSCs) and monocytic macrophage-derived suppressor cells (mMDSCs), whereas B16F10 and AP-3 tumors were poorly infiltrated by immune cells overall and therefore immunologically cold. Importantly, CT26 and RENCA had the most balanced population of immune cells including the highest CD4 + and CD8 + T cell populations of all the models indicating a robust immune reaction even in the absence of immunotherapy [ 71 ]. Lastly, they note that while these models may not clearly translate to the clinic, they do provide a platform to evaluate the ability of immunotherapeutic approaches to achieve responses in tumors with a different immunophenotypic background.
Finally, due to the abundance of well-established models and the advantage of an immunocompetent system, syngeneic models have been widely utilized as a pre-clinical tool for immunotherapy screening and it is thus important to characterize whether pharmacokinetic (PK) and pharmacodynamic (PD) parameters can be accurately translated into human clinical trials. Using the syngeneic MC38 tumor–bearing C57BL/6 mice, it was demonstrated that this model was useful for optimizing dose-range selection for the anti-PDL1 antibody pembrolizumab in early clinical development. As a result, they concluded that antibody distribution kinetics, drug association and dissociation, receptor occupancy, and dose response to a wide range of tumor growth rates were all measures that could be allometrically scaled to human parameters and accurately simulate findings in the clinical setting for selection of the lowest effective dose [ 77 ].
Drug discovery and drug screening
Xenograft and syngeneic mouse models have been extensively used as a means to assess the ability of conventional therapeutic agents to alter tumor growth or volume [ 56 , 60 , 78 – 86 ]. However, there appears to be divergence among xenograft and syngeneic mouse models in terms of their responsiveness to conventional chemotherapeutic agents. A retrospective based literature search comparing the responses of cell lines, xenograft, and syngeneic mice to thirty-one different cytotoxic cancer drugs found markedly distinct outcomes that differed across tumor types. For the four solid tumor types examined, colon, breast, ovarian, and NSCLC, they correlated the pre-clinical in vitro activity of each drug with phase II response rates by tumor type [ 56 ]. They also calculated whether the response in one tumor type could predict response in the same tumor type, in the other three tumor types combined, or in all four tumor types combined. In this case, it was found that cell lines could predict response in NSCLC, breast, and ovarian cancer, whereas syngeneic mouse models were not predictive and cell line–derived xenografts were predictive for NSCLC and ovarian cancer, but not breast cancer and colon cancer. In contrast, a screen of seven different syngeneic models of varying cancer types (two leukemia models and five solid tumors) found that syngeneic in vivo models can be used as a platform for drug screening, particularly for lymphoma, melanoma, and breast cancer [ 62 ].
Much like drug response, the predictive value of syngeneic models for assessing drug pharmacokinetic and pharmacodynamic parameters is dependent on the syngeneic model and tumor type being studied. In lymphoma syngeneic models, for example, the pharmacokinetic properties of rituximab plasma concentration and overall efficacy are significantly influenced by tumor burden in mice similar to what is encountered in human clinical studies [ 87 ].
However, when taken together, the predictive utility of this cell-derived xenograft and syngeneic mouse models for drug response differs significantly between cancer types, with some positive results in melanoma and lymphoma, and disappointing results in other cancer types such as colon, breast, ovarian, and lung cancer [ 62 , 63 ]. Further, even within the same cancer type, different syngeneic tumor cell lines have demonstrated markedly different molecular features and immunologic profiles in breast cancer, as well as renal and colon cancer [ 71 , 88 ]. An additional concern regarding this model system is the lack of an accurate tumor microenvironment with stromal, vascular, and immune components when using either flank or orthotopic injection [ 89 ]. Insofar as these specific cancer types are concerned, thorough interrogation of a particular syngeneic model regarding gene expression pattern and immunologic profile within the context of the human target population is required.
To further enhance the mechanistic and clinical relevance of pre-clinical in vivo models, GEMMs and PDXs models were developed.
Genetically engineered mouse models
Genetically engineered mice, or transgenic mice, were first described in the early 1980s following development of techniques that allowed stable transmission of genes to successive generations upon injection of cDNA into mice pronuclei [ 90 ]. This technique led to establishment of models of oncogenesis whereby oncogenes were overexpressed, or tumor suppressor genes silenced and this, in turn, yielded spontaneous tumor formation. The earliest studies with genetically engineered mouse models (GEMMs) demonstrated that inserting oncogenes such as ERG, KRAS and MYC, for example, into transgenic mice led to the development of cancer [ 91 , 92 ]. Thus, tumor development in these models was driven by genetic manipulation. As will be discussed, newer methods including Cre/loxP gene silencing, viral vectors, or CRISPR/Cas9 gene editing have emerged that dramatically alter the time scale of producing GEMMs [ 70 ]. GEMMs can also be induced to develop spontaneous tumors upon exposure to environmental factors (i.e., carcinogens, radiation) which can induce single nucleotide changes in genes and recapitulate a patient’s tumor [ 57 , 93 , 94 ].
Some of the notable limitations of GEMMs are that generating and propagating them requires dedicated labor, is time consuming, expensive, and as already discussed there is less resemblance to human tumors. However, GEMMs can play an important role in dissecting out specific molecular events in oncogenesis and in determining the relationship between these events and therapeutic responsiveness.
GEMMs are, arguably, a very effective mouse model for studying tumor heterogeneity and evolution, because they are genetically tractable, allowing investigation of specific mutations in a stable genetic background [ 57 ]. As noted, GEMMs are classically generated by inserting inducible or constitutively expressed cDNA, encoding tumor suppressor genes or oncogenes, into mice via direct injection of mouse oocytes or by means of viral vectors [ 57 , 95 ]. More recently, targeted disruption of genes using Cre/loxP recombinase has been developed that allows conditional expression or deletion of genes.
Niknarfs et al. used transgenic GEMMs to characterize clonal evolution in pancreatic cancer [ 96 ]. The two GEMMs, known as KPC (LSL-Kras G12D/+ ; LSL-Trp53 R172H/+ ; Pdx1-Cre) and KPTC (LSL-KRASG12D/ + ; LSL-Trp53R172H/ + ; Tgfbr2flox/ + ; Ptf1aCre/ + ) are conditional mice that when bred with cre-recombinase-expressing mice, lead to expression of active mutant Kras, and loss of p53 (by means of a dominant negative mutation) [ 96 ]. These mice closely mimic the histopathology and clinical features of PDAC and are more biologically relevant models for studying clonal evolution and tumor heterogeneity [ 96 ]. Based on their results, these authors concluded that KPC and KPTC mice accumulate sub-clonal somatic mutations as measured by copy number alterations in essential PDAC pathways including chromosomes containing the Cdkn2a gene (chr4), Tgfbr2 gene (chr9), and Trp53 (chr11) [ 96 ]. Other modifications in DNA damage response genes (Msh3), and genes involved in cellular recovery to DNA damage (Mastl) were also noted [ 96 ]. This group also discovered an unrecognized role for the gene Nlrp1b (part of a family of immune pattern recognition receptors) that was found to have undergone focal homozygous deletion in three mice (KPC8, KPC9, and KPTC26) [ 96 – 98 ]. Nlrp1b is also somatically altered in human pancreatic cancers. Ultimately, by modeling the genomic mutations observed in their models across different segments of the tumors, they could also assess the spatial heterogeneity within each of the mouse tumors allowing them to construct phylogenetic trees charting the evolutionary history of cells from each segment [ 96 ]. However, a liability of this method is that it can alter the germline. With more recent CRISPR/Cas9 techniques, researchers can move away from blunt germline alterations of gene expression to more controlled, targeted tissue-specific changes. Indeed, the use of CRISPR/Cas9 gene editing is now enabling manipulation of numerous cancer genes simultaneously [ 99 ].
As was discussed with organoids, single-cell analysis is an emerging tool that can be used in tandem with mouse models such as GEMMs to trace cell lineages with applications in cancer [ 100 , 101 ]. Also, the applications of these combinations of technologies can be used to address tumor heterogeneity in non-cancer tumor cells, such as stromal cells. For example, Bartoschek et al. used single-cell analysis to identify spatially and functionally distinct classes of cancer-associated fibroblasts from a MMTV-PyMT mouse breast cancer model [ 102 ].
Compared with other mouse models, GEMMs are well suited to investigating metastasis and there is a growing body of literature demonstrating de novo metastasis of breast and pancreatic cancer tumors [ 66 , 103 , 104 ]. This is highlighted in studies using the KPC pancreatic adenocarcinoma transgenic mouse model, which carries both inactivating p53 and activating KRas mutations [ 55 , 105 ]. As a result, these mice develop spontaneous tumors which metastasize to the liver at a high rate of 50–75% of mice [ 55 ]. In addition, in contrast to patient-derived xenograft or syngeneic mouse models, these mice also exhibit significant stromal and fibroblast infiltration, similar to that observed in human pancreatic adenocarcinomas [ 105 ]. These features make the KPC GEMMs useful models for studying the biology of metastasis, anti-metastatic agents, and treatment resistance [ 55 , 105 , 106 ]. For example, Morton et al. showed that the Src inhibitor could inhibit metastasis in KPC GEMMs [ 92 ]. There are a number of other GEMM models of PDAC with mutant p53 and Kras which also have fibrous desmoplastic stroma (that interferes with both drug penetration and immune infiltration), as well as PDAC GEMM models with PanIN cells that undergo epithelial-mesenchymal transition (EMT), further supporting their clinical relevance [ 9 , 107 – 109 ].
Finally, a number of recent studies have used GEMMs to delineate mechanisms of metastasis in small-cell lung cancer (SCLC) [ 110 – 112 ]. Building on the observations that p53 and RB are mutated in more than 90% of SCLCs, mouse models were established where these genes could be conditionally silenced using the Cre/loxP recombination [ 113 ]. Deletion of the cell cycle inhibitor, p130, in this model leads to enhanced tumor development [ 113 ]. Denny et al. used loxP-flanked Trp53 f/f , Rb1 f/f , p130 f/f mice crossed with R26 mT/mG mice to establish conditional knock-down transgenic mice that, when administered adenovirus containing the cre-recombinase via inhalation, were deficient for p53, Rb1, and p130 [ 110 , 114 ]. These mice, in turn, develop tumors that are tomato (mT) negative and GFP positive owing to the cre-mediated excision of mT in p53/Rb1/p130-deficient tumor cells, but not in normal cells [ 110 , 114 ]. In this way, it was possible to track and visualize GFP-positive tumor cells that metastasized. Using this system, the authors were able to identify a putative driver of metastasis, the transcription factor Nfib, which is involved in chromatin remodeling [ 110 ]. This same study also employed NSG mice to verify the metastatic potential of high and low Nfib levels via subcutaneous administration or transplantation of SCLC cells deficient in or overexpressing Nfib [ 110 ]. Other groups discovered a similar relationship between Nfib and SCLC metastasis using Trp53, Rb1 conditional knock-down GEMM mouse models [ 111 , 112 ]. These are just a few specific examples demonstrating that GEMMs may be a useful for studying metastasis.
GEMMs may also be utilized to study epigenetic regulation of metastasis. Recent studies show that epigenetic modification of immune cells can serve as a precondition for premetastatic microenvironment establishment and metastasis initiation [ 115 ]. These studies highlight how different mouse models can work in tandem to overcome limitations of any one model. For example, a combination of GEMMs, xenograft mice, and NSG mice were used to assess the effectiveness of epigenetic modifying drugs on myeloid-derived suppressor cells (MDSCs) in mediating metastasis [ 116 ]. First, syngeneic C57/BL/6 mice or BALB/c mice were either subcutaneously or orthotopically implanted with different cell lines known to aggressively metastasize to the lungs: Lewis lung carcinoma cells (LLC) (Sub-Q into C57/BL/6 mice), HNM007 esophageal squamous cell carcinoma cells (Sub-Q into C57/BL/6 mice), or 4T1 mammary cancer cells (orthotopic injection into mammary fat pads of BALB/c mice) [ 116 ]. Tumors were resected and metastasis was assessed by immunofluorescence and histology. Next, CD45.1 MDSCs were isolated from the LLC or HNM007 tumor–bearing mice and transplanted into mice congenic at the CD45 Ly5 locus (B.6SJL- Ptprc a Pepc b /BoyJ Ly5.1) [ 116 ]. The NSG mice transplanted with LLC tumor tissue were used as a model to guide dosing with the epigenetic modifying drugs azacitidine and entinostat. Host MDSCs derived from CD45.1 mice-bearing LLC tumors were then adoptively transferred into CD45.2 congenically marked mice via tail vein injection in order to track the primed tumor–derived MDSCs [ 116 ]. Treating these mice with low dose epigenetic modifying drugs led to a reduction in trafficking of MDSCs to premetastatic sites together with enhanced disease-free survival [ 116 ]. This was confirmed using B6.129S4- Ccr2 tm1Ifc /J mice, a knockout mouse lacking expression of the Ccr-2 gene, an important regulator of monocyte migration from the bone marrow to the tumor microenvironment [ 116 ]. Therefore, by leveraging the advantages of each of these mouse models, they demonstrated that epigenetic changes in MDSCs prime them to develop premetastatic niches for tumor cells even after tumor removal.
Another recent example focuses on the role of the epigenetic modifier polycomb repressor complex 2 (PRC1) in mediating stemeness, metastasis initiation, and local tumor immunosuppression in double negative prostate cancer (DNPC) [ 115 ]. A combination of mouse models was utilized including nude mice, NSG mice, Pten PC −/− FVB/NJ mice and Pten PC −/−Smad4 PC −/− FVB/NJ mice [ 115 ]. The nude mice were used as the model for metastasis due to the lack of an immune system. Nude mice were injected intracardially with luciferase-labeled androgen receptor (AR)–negative PC3M cells in the presence or absence of RNF2 (ring finger 2) shRNA (small hairpin RNA), a catalytic component of PRC1, thereby knocking down PRC1. Silencing RNF2 suppressed metastasis. The Pten PC −/− mice develop prostate cancer but have little to no metastasis, whereas the genetically modified Pten PC −/−Smad4 PC −/− develops metastatic prostate cancer [ 117 , 118 ]. This provides a model for studying how PRC1/RNF2 alters prostate cancer cells to influence metastasis. Briefly, shRNA-silenced prostate cancer cells from Pten PC −/−Smad4 PC −/− lacking functional PRC1 were injected into nude mice and metastasis to the bone and liver was reduced. Through a combination of RNA-seq and chromatin immunoprecipitation (CHIP) sequencing, they identified CCL2 as the target of PRC1 that mediates metastasis initiation and immunosuppression. CCL2 promotes recruitment of MDSCs and tumor-associated macrophages (TAMs) that create an immunosuppressed tumor microenvironment and favor bone colonization in prostate cancer [ 115 ]. Finally, FVB/NJ mice were inoculated with cells from their prostate cancer model ( Pten PC −/−Smad4 PC −/− ) and treated with a small-molecule inhibitor of RNF2, identified in a screen of a compound library–inhibited metastasis. Combination of the RNF2 inhibitor (GW-516) with anti-CTLA-4 and anti-PD-1 antibodies completely suppressed metastasis. Therefore, again, multiple mouse models can be used in tandem to study epigenetics, metastasis, and test novel compounds.
Lastly, an important caveat for congenic GEMM mouse strains that should be mentioned is that they appear to harbor significant amounts of passenger mutations as a consequence of genetic variation from embryonic stem cells (ESCs) used to establish the mouse lines [ 119 ]. While this likely poses less of an issue for the vast majority of translational cancer mouse studies, this could have the potential to interfere with interpretation of results in studies using congenic mice particularly as it relates to studies focused on tumor heterogeneity and identification of driver genes.
Like syngeneic mouse models, GEMMs have an intact immune system (albeit a mouse immune system) so they can more accurately model the interaction of tumors and the immune system in terms of tumor development and response to therapeutics [ 69 ]. This is in part due to the native development of the tumors in a microenvironment that adapts and changes with the tumor [ 89 ]. This microenvironment contains stromal elements, vasculature, and immune cells that influence the tumor’s relationship with the immune system [ 89 ] However, there are a number of limitations of using GEMMs in immune-tumor interaction studies that need to be considered [ 89 ]. Due to the fact that these mice are genetically modified, there can be substantial variability in the tumor genotype-phenotype penetrance and latency of tumor development [ 89 ]. Also, tumor monitoring and therapeutic response in GEMMs are, in large part, assessed by non-invasive imaging [ 89 ]. Consideration also needs to be paid to the consistency of immune targeting between the GEMM murine tumor model and the corresponding human tumor targets, which could affect clinical translation particularly for the development of immunotherapeutic vaccines [ 89 ]. However, there is a growing body of literature using GEMMs to study immune-tumor interactions ranging from T cell function, immunogenicity of tumors, and B cell contributions to tumor development and treatment response [ 70 , 120 ].
Single-cell analysis is also being used along with GEMMs to study immune-tumor interactions. Single-cell analysis has been used to explore the relationship between immune cell infiltration and tumor progression in prostate adenocarcinoma GEMMs deficient for Pten and Smad4 [ 121 ]. Loss of PTEN has been shown, in certain cancer models, to be associated with an immunosuppressive tumor microenvironment [ 89 ]. In their study, Wang et al. identified MDSCs as the major infiltrating immune cell in tumors from these mice, whose recruitment to tumors is driven by tumor production of the chemokine CXCL5. Single-cell analysis was performed on cells isolated from Pten pc −/− Smad4 pc −/− mouse blood, lymph, spleen, and primary tumors. The single cells were then immunophenotyped and a tree of cell types isolated from the aforementioned sources was established revealing a striking increase in MDSCs. They went on to identify the underlying signaling pathways mediated by Hippo-Yap1 in the altered expression of CXCL5 in these tumors. Inhibition of the CXCL5 receptor on MDSC or inhibition of YAP1 led to reduced migration of MDSCs to the site of the tumor [ 121 ]. Along these lines, there are a number of other recently published studies that have used single-cell techniques in tandem with GEMMS in detailing immune-tumor interactions in breast cancer and lung cancer [ 102 , 122 ].
GEMMs may serve as a useful platform for drug discovery, assessment of drug efficacy, and as a tool to assess multi-organ system adverse effects [ 55 , 70 ]. GEMMs can also be used to assess how existing and emerging drugs can alter the mutational profile of tumors and lead to treatment resistance [ 55 , 70 ]. For example, Mitrofanova et al. conducted a study using GEMMs to correlate expression levels of prostate cancer driver mutations such as FOXM1 and CENPF to those in human prostate cancer databases to predict drug response by targeting these specific mutational drivers [ 123 ]. They demonstrated that treatment-responsive genes modelled utilizing GEMMs can be used to identify patients that are likely to benefit from treatment with drugs that co-target specific pathways such as the MAP-kinase or mTOR signaling pathways [ 123 ]. In addition to targeting a specific signaling pathway, these models can also be used more widely to predict drug response to a number of different inhibitors from various classes. Chesi et al. performed a study in which they used Vk*MYC multiple myeloma transgenic mice to predict responses to six different classes of inhibitors, and demonstrated that drug response was indicative of clinical activity, with a positive predictive value of 67% in associated clinical trials [ 124 ]. The study also reported a negative predictive value of 86% for clinical inactivity, indicating that this model may also have translational value for prediction of both drug response and resistance [ 124 ].
To overcome some of the limitations of GEMMs, validate mutational changes and results of drug screens, GEMM models can be used in tandem with other mouse model systems such as PDX (discussed below). For example, Liu et al. recently described the development of a triple negative breast cancer (TNBC) GEMM [ 125 ]. They generated two cohorts of mice: a Brca1 -deficient line (K14 cre , Trp53 flox/flox , Brac1f lox/flox ) and a Brca-proficient wild-type line (K14 cre , Trp53 flox/flox , Brac1f wt/wt ) [ 125 ]. WES and RNAseq together with copy number alteration analysis of the TNBC tumors revealed several focal amplifications on chromosomes 6 and 9 involving the Met and Yap1 loci which corresponded to elevated mRNA levels for both [ 125 ]. They also found that several of the tumors expressed Fgfr2 and Raf1 fusion genes, both apparently products of chromosomal translocations. These genetic changes favored oncogenesis via corresponding changes in signal transduction pathways such as MAPK and PI3K, thus revealing therapeutic opportunities [ 125 ]. To test the response of these tumors to therapeutic agents, they transplanted the TNBC tumors into nude mice and assessed their response to several targeted drugs. In the case of tumors which spontaneously acquired the Fgfr2 or Raf1 fusion proteins, they tested an FGFR inhibitor NVP-BGJ398, the MEK inhibitor trametinib, or the MET inhibitor crizotinib [ 125 ]. They demonstrated that tumors with acquired Fgfr2 fusion proteins were initially responsive to single-agent FGFR inhibition with NVP-BGJ398 but developed resistance in three of six models with an average return to initial tumor volume of 43 days. They further went on to show that the combination of NVP-BGJ398 with Olaparib (a PARP inhibitor) resulted in complete responses with no relapse in all six of the models [ 125 ]. This study exemplifies how multiple models (cell culture, GEMM, and orthotopic nude mice) can be used in tandem to explore specific facets of tumor biology and response to treatment.
Beyond utilization as a simple screening platform for pharmacologic response, GEMMs have shown value for modelling drug pharmacokinetic and pharmacodynamic properties, and in some cancer sub-types may be superior to other in vivo models. Combest et al. assessed the pharmacokinetic properties of carboplatin in mouse melanoma models and compared results between PDX mice and GEMMs with those observed in the clinical setting [ 126 ]. Their findings demonstrate that although the carboplatin plasma PKs of each model were similar, the carboplatin concentration in tumors of the GEMMs more closely resembled those of melanoma patients as compared with the xenograft (A375) model to a significant degree, with a murine-to-human tumor extracellular fluid (ECF) drug concentration ratio of 0.13 and 0.86 for the xenograft or transgenic model, respectively. [ 126 ]. This example highlights the need for more studies than model pharmacokinetic drug properties in mouse tumor models.
Patient-derived xenograft mouse models
Patient-derived xenograft models were first created and published in 1969, when Rygaard and Polvsen first minced, and then injected a colonic adenocarcinoma sample from a 74-year-old patient into athymic nude mice [ 127 ]. This model, established for the first time more than five decades ago, has several distinct features when compared with cell line–derived xenografts and syngeneic or transgenic mouse models. Established as a useful tool for translational research, PDX models have the advantage of maintaining the cellular and histopathologic structure of the original tumor thus recapitulating the heterogeneity observed in patients [ 56 , 78 , 128 , 129 ]. This characteristic makes them a better tool compared with cells lines for studying drug efficacy and development: in fact, substantial limitations have been observed with conventional cell line–derived xenograft models for drug screening and evaluation of pre-clinical efficacy of drugs due to the loss of hallmarks, such as genetic and epigenetic alterations, resulting in minimal resemblance to the parental tumors [ 128 , 130 ].
PDX mice are created via subcutaneous or orthotopic implantation into an immunodeficient mouse, and in contrast to cell line–derived xenografts, they are not propagated on plastic. There is wide variation in engraftment rates and time-to-engraftment among different cancers that may be impacted by the method used to implant the tumor (i.e., subcutaneous, orthotopic, or kidney capsule) and mouse strains [ 78 , 128 , 131 , 132 ].
PDX are commonly used worldwide in pre-clinical trials for the development of anti-cancer drugs to support and validate the translation into clinical trials. In fact, many global PDX repositories have been generated and are currently available for pre-clinical research ( https://www.europdx.eu/ ; https://www.pdxfinder.org/ ; https://www.crownbio.com/ ; https://championsoncology.com/ ; https://www.jax.org/ ; http://www.pdx.dnalink.com/index ) [ 133 ].
The most recent application of these fine pre-clinical models have been “co-clinical trials”, where PDX, also known as “avatar” or “mirror” models, are generated using specimens derived from patients participating in the clinical trials and pre-clinical studies which are run in parallel, in real time, to the human trials. This approach has been effectively used in the past few years as a model for personalized medicine because they are able to longitudinally predict, with high accuracy, drug response, or resistance before these events can be observed in the donor patient [ 134 – 140 ].
Challenges and limitations of PDX models
The PDX mouse model is frequently cited as being highly representative of human tumors in terms of heterogeneity, clonal evolution, and response to treatment [ 81 , 131 , 141 – 145 ]. Recently, groups with wide expertise in the model have sought to assess the validity of PDX models in recapitulating different types of cancers and have highlighted many of the limitations and challenges of PDX models [ 128 , 131 , 146 ]. All groups working with PDX models argue that more studies should incorporate standardized validation tools to improve the reproducibility and to increase success rates of translational studies [ 72 , 78 , 79 , 131 , 132 , 146 ].
Among some of the main challenges is the gradual replacement of human stroma with mouse stroma. In fact, after few passages, tumor-associated stroma gets replaced with murine-derived ECM (extracellular matrix) and fibroblasts, causing changes in the paracrine regulation of the tumor that might interfere with drug distribution and effectiveness [ 147 ].
Another challenge is the route of implantation. There are open questions surrounding the most favorable route of administration indicating the need for validation studies to address the optimal implantation site. Although orthotopic models seem to better mimic metastatic cancer models, subcutaneous administration is more commonly used because it is easier to assess drug efficacy [ 138 ].
The time course of engraftment is also a formidable challenge and is a limiting factor in the use of PDX models for co-clinical trials. Some models take 4–8 months to establish and this is more than what patients can wait to start treatment. In order to overcome this issue, some groups are switching to the use of organoids models to evaluate potential treatment sensitivity [ 24 , 25 ].
Lastly, in order to establish standard PDX models, a key requirement is that the mice cannot have an intact immune system. This has impeded the use of PDX mice in studies assessing immune checkpoint–blocking agents [ 148 , 149 ]. This is also driven, in part, by gradual replacement of engrafted stromal cells (and immune cells found in the tumor) with mouse cells leading to a more murine-like tumor microenvironment [ 131 ]. For these reasons, the development of humanized PDX models where the immune system is reconstituted in the PDX-implanted mouse represents a potential advancement for researchers [ 149 , 150 ].
Nevertheless, despite these challenges, PDX models are considered among the most robust and clinically relevant models for drug screening and drug discovery. In fact, since 2016, the National Cancer Institute (NCI) has stopped using the NCI-60 panel (containing 60 human cancer cell lines) and switched to PDX models for anti-cancer drug screening [ 56 , 78 , 79 , 82 , 128 , 133 , 137 , 138 , 141 , 151 ].
As previously mentioned, PDX models present limitations for studying metastasis, particularly if subcutaneous transplantation is used [ 55 , 66 , 131 , 152 ]. The combination of the absence of an intact immune system and mouse stromal environment can influence disease progression and metastasis [ 66 , 153 ]. Recently, Sprouffske et al. assessed this issue and used a bioinformatics approach to investigate the genetic heterogeneity during breast cancer metastasis [ 152 ]. Using WGS, they discovered that the mouse stromal environment can confound interpretation of intra-tumor heterogeneity and that the method of developing the PDX metastasis models can influence genetic changes in occurring during metastasis [ 152 ]. For example, tail vein injection of breast cancer PDX metastasis models exhibits a loss of heterozygosity compared to PDX mice with orthotopically transplanted breast cancer tumors that develop metastasis [ 152 ]. Thus, although a liver metastasis–derived PDX will better recapitulate human tumors, both cell line–derived and PDX in vivo subcutaneous models are limited in their ability to simulate metastasis, and more studies are required to address this unmet need.
As noted several times, in addition to limitations in modeling metastasis, PDX models are immunocompromised as they are propagated in mice that lack a fully adaptive immune system. Thus, the influence of immune cells on tumor growth and response to treatment is poorly assessable in this model. However, there are several groups currently working to create mouse models with partial immune systems, primarily human T cells, to evaluate therapeutics used in the clinic, each which comes with challenges that will be discussed [ 149 ].
Several strains of mice can be used to generate PDX models. Traditional PDX models use athymic nude mice or severe combined immunodeficiency (SCID) mice [ 89 ]. Athymic nude mice lack T cells but still retain B cells as well as many elements of their innate immune response including NK cells and neutrophils [ 89 ]. Athymic mice are suitable hosts for human cancer cell lines, but NSG are more suitable for hosting human primary tumors [ 89 , 154 ]. SCID mice, on the other hand, have genetically impaired VDJ recombination leading to disruption in T and B cell development with commensurate deficiency in these critical adaptive immune system cells and therefore cannot be used to evaluate anti-PD1 or anti-CTLA4 immunotherapy agents [ 89 , 90 ]. In response to this, several groups are developing humanized PDX models reconstituted with human immune systems using novel approaches. These include reconstituting immunodeficient mice with mature immune cells (i.e., PBMCs or tumor-infiltrating lymphocytes) prior to transplanting patient tumor tissue containing human stromal cells along with any tumor-infiltrating human immune cells, while other groups have begun developing “humanized mice” reconstituted with human CD34 + hematopoietic stems cells (HSCs) following sub-lethal irradiation in immunodeficient mice, thereby repopulating them with a largely human immune system that includes B and T lymphocytes as well as myeloid cells [ 55 , 149 , 150 , 155 – 159 ].
The benefit of the PMBC method is the ability to transplant patient-matched immune cells from the blood into mice prior to tumor challenge thereby limiting antitumor immune effects derived from allogeneic responses due to HLA mismatching [ 131 ]. Furthermore, it was recently demonstrated that tumor-specific T cells can be found in the circulation of cancer patients and may be important for responses to PD1 blockade [ 160 ]. Previous studies have demonstrated improved overall engraftment that results in high chimerism of human lymphocytes using mice with deficiencies in both their adaptive and innate immune systems as recipients [ 161 ].
Yet, there are drawbacks. First, not all immune cells engraft equally as many murine-derived cytokines do not cross-react with human receptors. This is particularly true for myeloid cells and is currently being addressed by various methods that include exogenous injection of growth factors and expression of human FLT3L [ 162 ]. Furthermore, the cells that do engraft undergo important phenotypic changes following repopulation of lymphogenic hosts that also further skew immune composition and activation. Most importantly, this xenograft system is limited by the narrow window of time for therapeutic studies as mice invariably succumb as a result of elicitation of profound xenograft-versus-host disease (xeno-GVHD) [ 131 ]. Ongoing studies are currently working to overcome this by using MHC-null NOG mice as recipients that have successfully been used for immunotherapy studies [ 163 ].
Another approach to overcoming severe xenograft versus host responses is transferring human CD34 + HSCs into irradiated, immunodeficient mice. As opposed to models with PBMCs containing mature T cells, the lymphocytes in the HSC transplant model develop in the murine host and therefore do not attack the host as a result of negative selection of thymocytes against murine MHC-peptide complexes [ 164 ]. In this system, human-derived thymocytes develop in the hosts’ thymus that are dominated by murine MHC-peptide complexes on thymic epithelial cells (TECs), which are important for both positive and negative selection [ 165 ]. Although human MHC-peptide complexes derived from donor immune cells are also present in the thymus, it is currently unknown what fraction of mature T cells are restricted to human versus mouse MHC peptides. Thus, counterintuitively, this model may initiate xenograft responses against human tumors upon challenge due to the absence of human MHC and peptides during negative selection. One way groups are attempting to overcome this issue is by introducing HLA genes in MHC-null mice that result in HLA-restricted T cells, albeit primarily HLA presenting murine rather than human peptides [ 166 , 167 ]. Currently, this approach is limited to a specific HLA gene and cannot match the complete HLA haplotype of the patients’ tumor. Lastly, other groups have also demonstrated that implanting human thymuses in HSC-reconstituted mice helps select for HLA-restricted T cells and ensure proper negative selection against human peptides [ 168 ]. In summary, there are well-designed experiments ongoing to develop and refine PDX models for use in testing immunotherapy, but it is unlikely that any of them will be feasible for large scale use or high throughput screening.
Greater emphasis has recently been devoted to the establishment of humanized PDX models in order to investigate the tumor and immune compartment effects of treatment as well as the interplay between these two systems. Indeed, PDX models show convincing evidence for their ability to model drug response in multiple human cancer types including breast cancer, ovarian cancer, SCLC, adrenal, and CRC [ 56 , 149 , 150 , 169 ]. There are a number of humanized mouse models with human-derived xenograft tumors as well as human immune systems that are currently in development and in use for drug screening and modeling of various oncologic diseases [ 170 ]. These models include bladder cancer humanized mouse models using NSG mice injected with CD34 + hematopoietic cells, breast cancer models created with NSG mice intrahepatically engrafted with breast carcinoma cell lines and engrafted with functional human immune systems, and CRC models via Rag2 −/− y c − / − mice injected with human PBMC’s and subcutaneously engrafted on the flank with CRC cell line HT-29 [ 171 , 172 ]. These systems are promising models of immune system and tumor interactions, as with the humanized breast cancer model created by Wege et al., where human immune cells are able to traffic and infiltrate the microenvironment and enable human tumor-immune system interactions to be studied [ 171 ]. In summary, humanized oncological models may address vital questions on tumor-immune system interactions, mechanisms of tumor escape, and therapeutic potential of immune modulation, and may be of significant importance in predicting response to immunotherapy such as checkpoint inhibition in the future [ 171 , 173 ].
To date, perhaps the most significant role for PDX in translational cancer research is in assessing drug response and translating in vivo drug screening data to the clinic [ 78 , 128 , 131 ]. A recent systematic review of retrospective studies correlated patient response in multiple oncologic indications with PDX response in vivo for cancer types including breast, ovarian, and small-cell lung cancer [ 169 ]. In support of the predictive capabilities of PDX models, they note a study that used a panel of seven human breast cancer patient-derived orthotopic xenografts (PDOXs) to predict patients’ responses to the chemotherapeutics docetaxel and 5-FU given in combination therapy with the monoclonal antibody trastuzumab, revealing an overall concordance in five of seven patients and corresponding PDOX models [ 169 ]. In the case of ovarian cancer, another study found that 19 of 21 PDXs exhibited congruent results of sensitivity or resistance with patients in retrospective studies with cisplatin treatment [ 169 ]. Lastly, in SCLC, strong correlations between the response rate to chemotherapy with cisplatin and etoposide combination treatment between SCLC patients and PDXs were demonstrated in seven of nine patient PDOX pairs [ 169 ]. While this systematic review highlights the potential role of PDX models in predicting tumor drug responses in certain cancer types such as breast cancer, ovarian cancer, and SCLC, primary research utilizing larger cohorts in other cancer types has additionally yielded interesting results. For example, Bertotti et al. produced a large cohort of patient-derived xenografts from 85 patients with metastatic colorectal cancer and characterized response to cetuximab (an anti-EGFR antibody) in correlation to patients in clinic [ 56 ]. In this panel of CRC patients and corresponding PDXs, all 85 were concordant regarding treatment response or resistance to cetuximab treatment. For treatment with cetuximab, the response rate (11%), disease stabilization (30%), or progression (59%) was in line with the data reported in the prospective analysis of the patients. Importantly, metastatic CRC xenografts retained the morphologic characteristics of the corresponding patient’s tumor, and serial mouse passaging did not substantially alter the genetic makeup of tumors as it related to copy number changes and hotspot oncogenic mutations. This indicates that in addition to being of benefit for prediction of drug response in CRC, PDX mouse models also retain key features of human tumors despite passaging and mouse stromal invasion [ 56 ]. However, the validity of PDX models as predictive tools in translational research has been questioned suggesting more validation tools are needed as part of PDX studies [ 146 ].
In modelling of drug pharmacokinetic and dynamic properties, PDX models have demonstrated promise in some systems while exhibiting limitations in others. Wong et al. carried out a retrospective PK/PD analysis of clinical response data from 8 well-characterized cytotoxic agents including 5-FU and docetaxel in comparison with response and PK/PD parameters in corresponding xenograft models with these same agents [ 174 ]. It was observed that regimens of docetaxel that were varied by dose and cycle duration in metastatic breast cancer models, such as the Cal51x1.1s PDX exhibited similar parameters of tumor growth inhibition and overall response to those documented in the clinical setting. Likewise, modelling of PK/PD parameters for treatment of CRC with 5-FU demonstrated that continuous infusion exhibited superior performance in comparison with a 5-day regimen in the Colo205 PDX model and was representative of clinical response in both cases [ 174 ]. Meanwhile, previous studies by Combest et al. concluded that genetically engineered transgenic mouse models may more closely recapitulate drug pharmacokinetic properties in comparison with PDX models in the setting of melanoma [ 126 ]. The limited pharmacokinetic data that exists for direct comparison of PDX and GEMM systems demonstrate that GEMM models may be superior in accurately modelling drug PK parameters in some cancer types [ 126 ].
Each of the tools discussed here plays important roles in unraveling tumorigenesis, identifying drug targets, and ascertaining drug efficacy (Fig. (Fig.1). 1 ). Two-dimensional cell culture still has a prominent role in cancer research and a long history of generating valuable results on underlying genetic changes in cancer that has led to the discovery of drug targets and therapeutic agents. Indeed, cell culture is a simple and cheap means to screen compounds and candidate drugs prior to more elaborate and predictive in vivo models. However, on its own, cell culture is subject to such profound shifts in gene expression with prolonged culture that its translational value is limited. The principle uses of cell culture are for preliminary drug screening/drug discovery (Fig. (Fig.1). 1 ). Although there has been work on cancer immunology using cell culture, there are better systems available as noted. Likewise, cell culture is not ideally suited for the study of metastasis, and while there are migration, invasion, and metastasis assays, they lack the robustness of mouse models.
Organoids are a more elaborate form of cell culture that can be used to study tumor heterogeneity (Fig. (Fig.1). 1 ). Organoids also appear to be effective and predictive platforms for drug screening and discovery, more so than cell culture but less than some mouse models (Fig. (Fig.1). 1 ). Similar to cell culture, organoids are not well suited for studying cancer immune function or metastasis.
Comparison of in vivo mouse model systems including xenograft, syngeneic, GEMMs, and PDX models reveals that there are unique benefits and limitations to each (Fig. (Fig.1). 1 ). Of these, cell line–derived xenograft mouse models appear to be the least valuable for studying tumor heterogeneity, tumor evolution, and immune-tumor interactions, but have demonstrated some success in studying metastasis and drug action (Fig. (Fig.1). 1 ). While syngeneic models offer a competent immune system, as discussed, particular care needs to be taken when choosing the murine tumor cell line used. Significant differences in genetic composition and immunologic profile have been demonstrated in this model within the same cancer type, and drug response results have been shown to differ between syngeneic models and human subjects for multiple indications [ 63 , 65 , 71 ]. Further concern regarding this model system is the lack of similar stromal and immune compartments in the tumor microenvironment following injection of tumor cells [ 89 ]. Although not without limitations, GEMMs appear to be a reasonable option for studying tumor heterogeneity, tumor evolution, metastasis, and to a lesser degree, immune-tumor interactions (with a mouse immune system). Although GEMMs also have predictive value in modelling drug response and PK parameters, they are still mouse tumors [ 123 , 126 ]. PDX mouse models remain the strongest in vivo model for predicting drug response in patients [ 128 ] (Fig, (Fig,1). 1 ). However, limited data for direct comparison of PDX and GEMM systems regarding PK modelling has demonstrated PDX tumors may be inferior in accurately recapitulating drug PK parameters in some cancer types [ 126 , 175 ], and further studies are needed. As noted, the lack of an immune system hampers the use of PDX mice in studying immune-tumor interactions or the effects of immunotherapy agents. However, the advent of humanized PDX mice has helped to fill this gap (Fig. (Fig.1 1 ).
Ultimately though, it is through the use of combinations of models that the ideal system may come closest to realization. Combining translational models is now frequently employed to leverage the advantages each system has to offer. The work described earlier by Lu et al., for example, typifies this translational combinatorial approach. Here, they describe how both syngeneic mouse models, which spontaneously develop tumors that aggressively metastasize to the lungs, were used in tandem with NSG mice transplanted with these tumors for drug screening [ 116 ]. Furthermore, coupling in vivo models with single-cell analysis technology and computational assays also stands to extend the resolution of data gleamed from any one technique.
Finally, there are a number of promising approaches being developed that extend beyond the scope of the current review and into clinical cancer diagnosis, but merit consideration given their potential use in pre-clinical cancer research. Advancements in imaging and spectroscopy have led to refinement of technology such as Raman spectroscopy and mass spectrometry for use in cancer detection and diagnosis [ 176 , 177 ]. Thus, it is conceivable that these technologies could be used in tandem with mouse models, such as PDX mice, to detect metastatic tumor formation and circulating tumor cells thereby extending the role of these models in cancer drug development.
Authors’ contributions
Michael W. Lee, Mihailo Miljanic, and Anna Capasso: conceptualization, writing-original draft preparation, writing-editing and reviewing. Todd Triplett: writing-original draft preparation, writing-editing and reviewing. Craig Ramirez: writing-editing and review. Kyaw L. Aung: writing-editing and review. S Gail Eckhardt: writing-editing and Reviewing
This work was financially supported by CPRIT grant RR160093 (S.G.E “CPRIT SCHOLAR IN CANCER RESEARCH) and the DoD grant #12935262 (awarded to Anna Capasso). The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Compliance with ethical standards
The authors declare that they have no conflicts of interest.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
COMMENTS
Technological advancements in cancer diagnostics
Cancer Treatment Research - NCI
At present, three main techniques are used in structural biology research: X-ray crystallography, nuclear magnetic resonance (NMR), and cryo-EM. 29 X-ray crystallography reveals atomic details for ...
New approaches and procedures for cancer treatment
The assay is now used in a wide range of applications—from ancestry reports and cancer research, to NIH's All of Us Research Program, and even to analyzing a plant's genome to see what influences insect and drought resistance. Learn more about how NCI partners with small businesses to advance innovations in cancer research and care.
Innovative approaches for cancer treatment: current ...
The future of early cancer detection
Nature Reviews Cancer 21, 747-752 (2021) Cite this article. Artificial intelligence and machine learning techniques are breaking into biomedical research and health care, which importantly ...
How CRISPR Is Changing Cancer Research and Treatment
Abstract. Clinical research is essential to introduce new treatments for cancer patients. Traditionally, a clinical research program starts with phase I trials, with the aim of describing the safety and identifying the dose and schedule to be tested in the subsequent phases of development. Phase II trials describe the antitumor activity of the ...
In clinical cancer research, proteomics techniques fall into two categories, proteomics expression and the other used in functional proteomics. The major challenge in proteomics is the availability of samples and purity of samples [51]. The following are major techniques used in proteomics: 5.1. Mass spectrometry (MS)
Research On Cancer
We use molecular epidemiology to examine the relationship of genetic and environmental risk factors to cancer etiology. Using laboratory techniques, investigators look for biomarkers of disease and use them to understand the underlying mechanisms of disease in populations. ... Read more about research on the Psychosocial Effects of Cancer ...
The field of cancer treatment has evolved significantly over the last decade with the emergence of next-generation therapeutic antibodies. Conventional treatments like chemotherapy pose significant challenges, including adverse side effects. Monoclonal antibodies have paved the way for more targeted and effective interventions. The evolution from chimeric to humanized and fully human ...
NGS-based cancer sequencing methods have expanded our understanding of cancer development, regulation, and progression to unlock new pathways for research. These techniques help detect changes in the cancer genome and identify their impact on the transcriptome, epigenome, and proteome. Unlike other methods, such as PCR and Sanger sequencing ...
Cancer Research. Due to disease complexity, cancer research is a multidisciplinary field that spans basic, translational, and clinical research. Advancements depend on an increased understanding of the tumor cell environment and cancer biological, etiologic, clinical, and genetic heterogeneity. Insights into the causes and mechanisms of cancer ...
The research teams used PET to identify dysfunctional biological pathways in 12 different types of cancer. They discovered numerous pathways associated with individual cancers as well as several that signaled either a high or low risk of cancer progression, effectively acting as potential biomarkers of outcomes.
Specifically, pathology data are used in prostate cancer, breast and colorectal cancer research utilize genomic data, and lung cancer is largely dependent on imaging data, especially CT scans. Despite those slight propensities per cancer type, we observe that all ML techniques, and especially DL techniques, are primarily used for the analysis ...
The new AI system, described Sept. 4 in Nature, goes a step beyond many current AI approaches to cancer diagnosis, the researchers said.(DOI 10.1038/s41586-024-07894-z) Current AI systems are ...
Pancreatic ductal adenocarcinoma (PDAC) is frequently detected in late stages, which leads to limited therapeutic options and a dismal overall survival rate. To date, no robust method for the detection of early-stage PDAC that can be used for targeted screening approaches is available. Liquid biopsy allows the minimally invasive collection of body fluids (typically peripheral blood) and the ...
In medical applications, zero-shot methods have been used to summarize unstructured radiology reports 106, ... In cancer research and clinical diagnostics, it seems intuitive that enriching image ...
Cancer of the thyroid has become the fastest-growing cancer among women in the past several decades. This study is aimed at using scientometric methods to identify research frontiers and development trends in the field of thyroid cancer (TC) research. We used the Scopus database to collect articles and reviews related to TC in November 2022.
This new method, combined with machine learning, accurately identifies cancer cells and could greatly improve breast cancer treatment and patient care. As breast cancer remains a leading cause of cancer-related deaths in women, innovations like these are crucial in improving treatment and overall patient care. Learn more about the method.
Biopsy techniques used in human cancer research showing promise in mussel testing ... St-Pierre has been a cancer researcher for over 20 years and first experimented with the approach of applying cancer-detection techniques to marine life a decade ago on a visit to the Kerguelen Islands, a remote archipelago over 1,000 miles off the coast of ...
Despite this, it is important that research continues. Technology is developing at a rapid pace. With this development comes the use of radio waves in different ways using different frequencies.
The University of Cincinnati Cancer Center's Eric Vick, MD, PhD, has been awarded a nearly $215,000 grant from the Leukemia and Lymphoma Society (LLS) and a $50,000 American Society of Clinical Oncology (ASCO) Young Investigator Award to continue research into a combination therapy treatment for acute myeloid leukemia (AML).
In 2011, the International Agency for Research on Cancer (IARC) classified radio wave exposure as a possible carcinogen to humans. The meaning of this classification was largely misunderstood and ...
Abstract. Recent developments in pre-clinical screening tools, that more reliably predict the clinical effects and adverse events of candidate therapeutic agents, has ushered in a new era of drug development and screening. However, given the rapid pace with which these models have emerged, the individual merits of these translational research ...
Abstract. Background: We examined the association between perceived discrimination and adherence to breast, cervical, and colorectal cancer screening recommendations among residents in a South Florida cancer center catchment area (n = 716).. Methods: Perceived discrimination was measured using the nine-item 'Everyday Discrimination Scale' and operationalized into quartiles (frequency of ...
Most U.S. polling organizations that conducted and publicly released national surveys in both 2016 and 2022 (61%) used methods in 2022 that differed from what they used in 2016. And change has continued since 2022. ... Research has found that Trump is popular among people who tend to sit out midterms but turn out for him in presidential ...